Journal of Inorganic Materials ›› 2021, Vol. 36 ›› Issue (10): 1047-1052.DOI: 10.15541/jim20210078
• RESEARCH ARTICLE • Previous Articles Next Articles
HE Junlong1( ), SONG Erhong2(), WANG Lianjun1(), JIANG Wan1
), SONG Erhong2(), WANG Lianjun1(), JIANG Wan1
Received:2021-02-05
Revised:2021-03-02
Published:2021-10-20
Online:2021-03-15
Contact:
SONG Erhong, associate professor. E-mail: ehsong@mail.sic.ac.cn; WANG Lianjun, professor. E-mail: wanglj@dhu.edu.cn
About author:HE Junlong(1996-), Master candidate. E-mail: woaichenzy@outlook.com
Supported by:CLC Number:
HE Junlong, SONG Erhong, WANG Lianjun, JIANG Wan. DFT Calculation of NO Adsorption on Cr Doped Graphene[J]. Journal of Inorganic Materials, 2021, 36(10): 1047-1052.
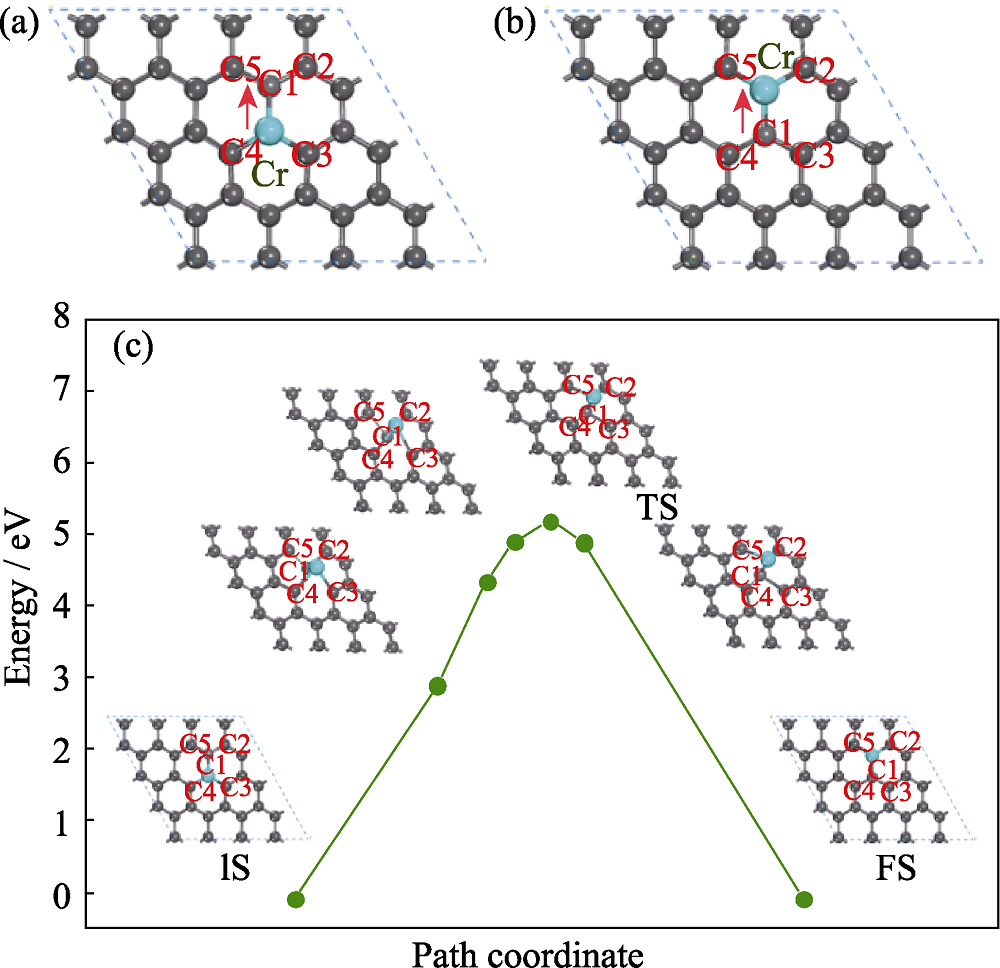
Fig. 1 Optimized structure of Cr-doped graphene (a) Initial state (IS); (b) Relaxed configuration of final state (FS); (c) Diffusion period of Cr atom on graphene; Gray and cyan spheres denote C and Cr atoms, respectively

Fig. 2 Atomic configuration of NO adsorption on graphene and Cr-doped graphene Graphene with N-end model (a) and O-end model (b), and Cr-doped graphene with N-end (c) and O-end model (d); Gray, cyan, blue, and red spheres denote C, Cr, N, and O atoms, respectively
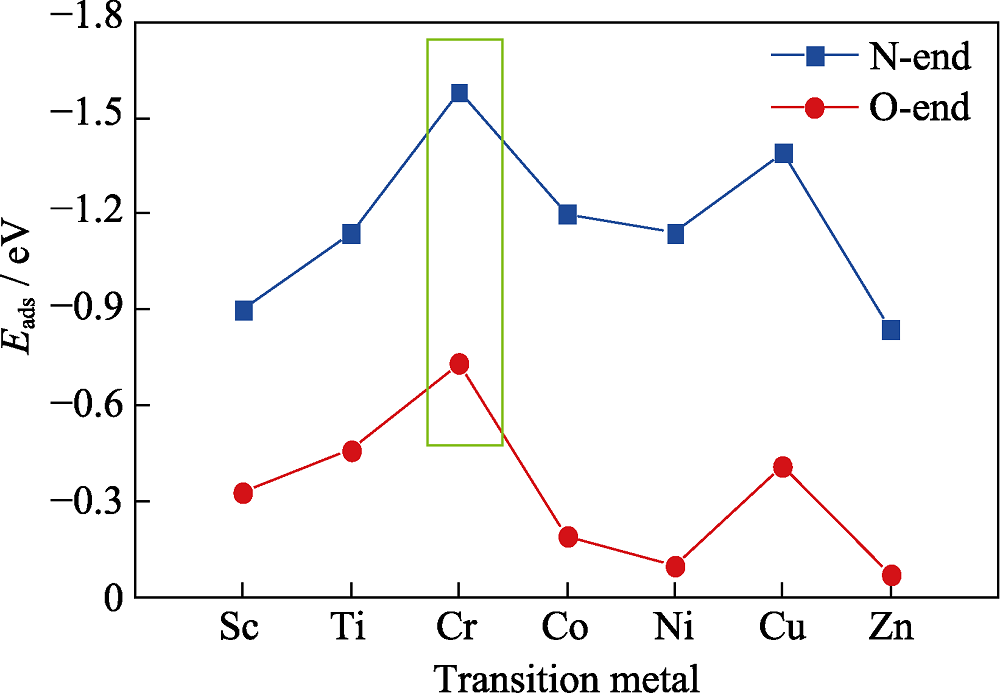
Fig. 3 Adsorption energy of NO adsorbed on 3d transition metal doped graphene via N-end and O-end model, respectively
| System | NO-O-end | NO-N-end |
|---|---|---|
| Graphene | -0.012 e | -0.009 e |
| Cr doped graphene | -0.119 e | -0.143 e |
Table 1 Charge change (∆Q) of graphene and Cr doped graphene after NO adsorption
| System | NO-O-end | NO-N-end |
|---|---|---|
| Graphene | -0.012 e | -0.009 e |
| Cr doped graphene | -0.119 e | -0.143 e |
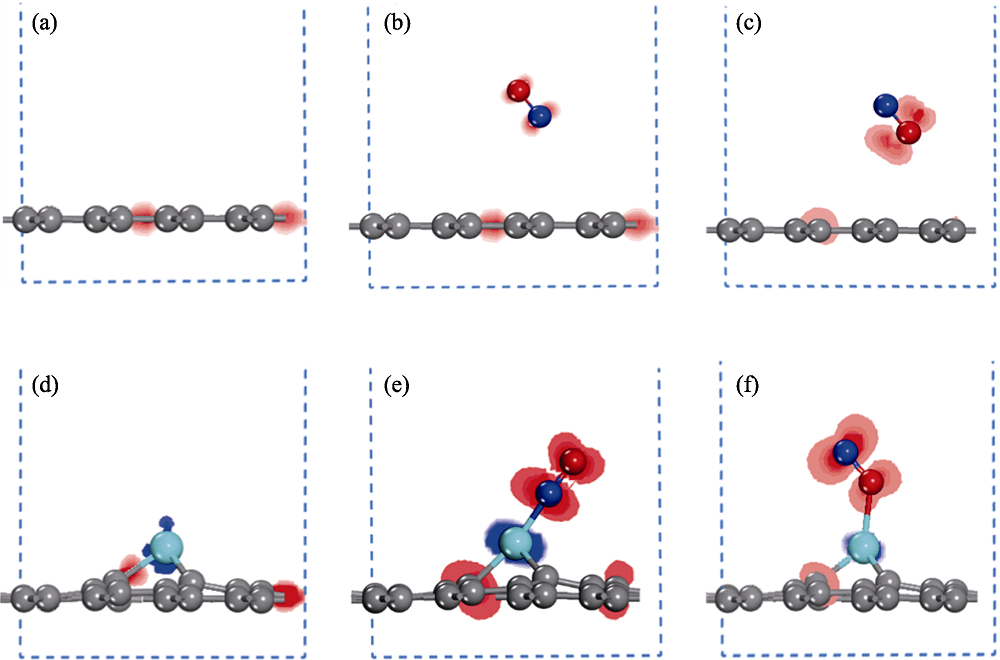
Fig. 4 Charge density difference of graphene and Cr-doped graphene before and after NO adsorption (a) Graphene; NO adsorption on graphene via (b) N-end and (c) O-end model; (d) Cr-doped graphene; NO adsorption on Cr doped graphene via (e) N-end and (f) O-end model; Red and blue regions represent accumulation and loss of electrons, respectively

Fig. 5 Density of states (DOS) of intrinsic graphene (a, b) and Cr-doped graphene (c, d) before (a, c) and after (b, d) NO adsorption
| [1] |
JOHNSON C, HENSHAW J, MCLNNES G. Impact of aircraft and surface emissions of nitrogen oxides ontropospheric ozone and global warming. Nature, 1992, 355(6355):69-71.
DOI URL |
| [2] |
LELIEVELD J, KLING MÜ, LLER K, et al. Effects of fossil fuel and total anthropogenic emission removal on public health and climate. Proceedings of the National Academy of Sciences, 2019, 116(15):7192-7197.
DOI URL |
| [3] |
POTYRAILO R A, GO S, SEXTON D, et al. Extraordinary performance of semiconducting metal oxide gas sensors using dielectric excitation. Nature Electronics, 2020, 3(5):280-289.
DOI URL |
| [4] |
TSAI Y T, CHANG S J, JI L W, et al. High sensitivity of NO gas sensors based on novel Ag-doped ZnO nanoflowers enhanced with a UV light-emitting diode. ACS Omega, 2018, 3(10):13798-13807.
DOI URL |
| [5] |
LI QIANG, SHI WANYAN, ZHANG CHEN, et al. SO2 non- equilibrium gas sensor based on Na3Zr2Si2PO12 solid electrolyte. Journal of Inorganic Materials, 2018, 33(2):229-236
DOI URL |
| [6] |
JIN W, HO H L, CAO Y C, et al. Gas detection with micro- and nano-engineered optical fibers. Optical Fiber Technology, 2013, 19(6 Part B):741-759.
DOI URL |
| [7] | LI J, YAN H, DANG H, et al. Structure design and application of hollow core microstructured optical fiber gas sensor: a review. Optics & Laser Technology, 2021, 135:106658. |
| [8] |
AKSHYA S, JULIET A V. A computational study of a chemical gas sensor utilizing Pd-rGO composite on SnO2 thin film for the detection of NOx. Scientific Reports, 2021, 11(1):970.
DOI URL |
| [9] | HANG T, WU J, XIAO S, et al. Anti-biofouling NH3 gas sensor based on reentrant thorny ZnO/graphene hybrid nanowalls. Microsystems & Nanoengineering, 2020, 6:41. |
| [10] |
CHU Y X, LIU H R, YAN S. Preparation and gas sensing properties of SnO2/NiO composite semiconductor nanofibers. Journal of Inorganic Materials, 2021, 36(9):950-958.
DOI URL |
| [11] |
KRENO L E, LEONG K, FARHA O K, et al. Metal-organic framework materials as chemical sensors. Chemical Reviews, 2012, 112(2):1105-1125.
DOI URL |
| [12] |
LEE J S, KWON O S, PARK S J, et al. Fabrication of ultrafine metal-oxide-decorated carbon nanofibers for DMMP sensor application. ACS Nano, 2011, 5(10):7992-8001.
DOI URL |
| [13] | GUO X, WANG X, YANG R, et al. EDTA assistant preparation and gas sensing properties of Co3O4 nanomaterials. Journal of Inorganic Materials, 2020, 35:1215-1221. |
| [14] |
LIANG JIRAN, ZHANG YE, YANG RAN,et al Room- temperature NH3 gas sensing property of VO2(B)/ZnO hierarchical heterogeneous composite with nanorod structure. Journal of Inorganic Materials, 2018, 33(12):1323-1329.
DOI URL |
| [15] |
XU SHUANG, YANG YING, WU HONGYUAN, et al. Preparation of one-dimensional Pt/SnO2 nanofibers and NOx gas-sensing properties. Journal of Inorganic Materials, 2013, 28(6):584-588.
DOI URL |
| [16] |
HAN SHUANGSHUANG, LIU LIYUE, SHAN YONGKUI, et al. Research of graphene/antireflection nanostructure composite transparent conducting films. Journal of Inorganic Materials, 2017, 32(2):197-202.
DOI URL |
| [17] |
NAN HUI, WANG WENLI, HAN JIANHUA, et al. Low-cost preparation of graphene papers from chemical reduction with FeI2/Ni2+ for conductivity and catalytic propert. Journal of Inorganic Materials, 2017, 32(9):997-1003.
DOI URL |
| [18] |
YANG G, LEE C, KIM J, et al. Flexible graphene-based chemical sensors on paper substrates. Physical Chemistry Chemical Physics, 2013, 15(6):1798-1801.
DOI URL |
| [19] |
CHOI J H, LEE J, BYEON M, et al. Graphene-based gas sensors with high sensitivity and minimal sensor-to-sensor variation. ACS Applied Nano Materials, 2020, 3(3):2257-2265.
DOI URL |
| [20] | YUAN W, SHI G. Graphene-based gas sensors. Journal of Materials Chemistry A, 2013, 1(35):10078-10091. |
| [21] |
XING WEIWEI, ZHANG CHENXIAO, FAN SHANGCHUN, et al. Research progress on resonant characteristics of graphene. Journal of Inorganic Materials, 2016, 31(7):673-680.
DOI URL |
| [22] |
PUMERA M, AMBROSI A, BONANNI A, et al. Graphene for electrochemical sensing and biosensing. TrAC Trends in Analytical Chemistry, 2010, 29(9):954-965.
DOI URL |
| [23] |
GOMEZ DE ARCO L, ZHANG Y, SCHLENKER C W, et al. Continuous, highly flexible, and transparent graphene films by chemical vapor deposition for organic photovoltaics. ACS Nano, 2010, 4(5):2865-2873.
DOI URL |
| [24] |
CASTRO NETO A H, GUINEA F, PERES N M R, et al. The electronic properties of graphene. Reviews of Modern Physics, 2009, 81(1):109-162.
DOI URL |
| [25] |
RATINAC K R, YANG W, RINGER S P, et al. Toward ubiquitous environmental gas sensors-capitalizing on the promise of graphene. Environmental Science & Technology, 2010, 44(4):1167-1176.
DOI URL |
| [26] |
NOVOSELOV K S, GEIM A K, MOROZOV S V, et al. Electric field effect in atomically thin carbon films. Science, 2004, 306(5696):666-669.
DOI URL |
| [27] |
MEYER J C, GEIM A K, KATSNELSON M I, et al. The structure of suspended graphene sheets. Nature, 2007, 446(7131):60-63.
DOI URL |
| [28] |
LEE C, WEI X, KYSAR J W, et al. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science, 2008, 321(5887):385-388.
DOI URL |
| [29] |
WANG LIN, TIAN LINHAI, WEI GUODONG, et al. Epitaxial growth of graphene and their applications in devices. Journal of Inorganic Materials, 2011, 26(10):1009-1019.
DOI URL |
| [30] |
WANG X, BAI H, SHI G. Size fractionation of graphene oxide sheets by pH-assisted selective sedimentation. Journal of the American Chemical Society, 2011, 133(16):6338-6342.
DOI URL |
| [31] |
XU Y, SHI G. Assembly of chemically modified graphene: methods and applications. Journal of Materials Chemistry, 2011, 21(10):3311-3323.
DOI URL |
| [32] |
WANG GUIXIN, PEI ZHIBIN, YE CHANGHUI. Inkjet- printing and performance investigation of self-powered flexible graphene oxide humidity sensors. Journal of Inorganic Materials, 2019, 34(1):114-120.
DOI URL |
| [33] |
SCHEDIN F, GEIM A K, MOROZOV S V, et al. Detection of individual gas molecules adsorbed on graphene. Nature Materials, 2007, 6(9):652-655.
DOI URL |
| [34] | HWANG E H, ADAM S, DAS SARMA S. Transport in chemically doped graphene in the presence of adsorbed molecules. Physical Review B, 2007, 76(19):195421. |
| [35] | ROMERO H E, JOSHI P, GUPTA A K, et al. Adsorption of ammonia on graphene. Nanotechnology, 2009, 20(24):245501. |
| [36] | CHEN C W, HUNG S C, YANG M D, et al. Oxygen sensors made by monolayer graphene under room temperature. Applied Physics Letters, 2011, 99(24):243502. |
| [37] |
YU K, WANG P, LU G, et al. Patterning vertically oriented graphene sheets for nanodevice applications. The Journal of Physical Chemistry Letters, 2011, 2(6):537-542.
DOI URL |
| [38] |
RUMYANTSEV S, LIU G, SHUR M S, et al. Selective gas sensing with a single pristine graphene transistor. Nano Letters, 2012, 12(5):2294-2298.
DOI URL |
| [39] |
AO Z M, YANG J, LI S, et al. Enhancement of CO detection in Al doped graphene. Chemical Physics Letters, 2008, 461(4):276-279.
DOI URL |
| [40] |
ZHENG Z, WANG H. Different elements doped graphene sensor for CO2 greenhouse gases detection: the DFT study. Chemical Physics Letters, 2019, 721:33-37.
DOI URL |
| [41] | KAUSHAL S, KAUR M, KAUR N, et al. Heteroatom-doped graphene as sensing materials: a mini review. RSC Advances, 2020, 10(48):28608-28629. |
| [42] |
SRIVASTAVA S, JAIN S K, GUPTA G, et al. Boron-doped few- layer graphene nanosheet gas sensor for enhanced ammonia sensing at room temperature. RSC Advances, 2020, 10(2):1007-1014.
DOI URL |
| [43] | ZHANG X, LU Z, TANG Y, et al. A density function theory study on the NO reduction on nitrogen doped graphene. Phys.Chem.Chem.Phys., 2014, 16(38): 20561-1-9. |
| [44] | PRAMANIK A, KANG H S. Density functional theory study of O2 and NO adsorption on heteroatom-doped graphenes including the van der Waals interaction. The Journal of Physical Chemistry C, 2011, 115(22):10971-10978. |
| [45] |
LIAO Y, PENG R, PENG S, et al. The adsorption of H2 and C2H2 on Ge-doped and Cr-doped graphene structures: a DFT study. Nanomaterials, 2021, 11(1):231.
DOI URL |
| [46] |
DELLEY B. An all-electron numerical method for solving the local density functional for polyatomic molecules. The Journal of Chemical Physics, 1990, 92(1):508-517.
DOI URL |
| [47] |
DELLEY B. From molecules to solids with the DMol3 approach. The Journal of Chemical Physics, 2000, 113(18):7756-7764.
DOI URL |
| [48] |
PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple. Physical Review Letters, 1996, 77(18):3865-3868.
DOI URL |
| [49] | DELLEY B. Hardness conserving semilocal pseudopotentials. Physical Review B, 2002, 66(15):155125. |
| [50] |
HIRSHFELD F L. Bonded-atom fragments for describing molecular charge densities. Theoretica Chimica Acta, 1977, 44(2):129-138.
DOI URL |
| [51] |
HENKELMAN G, JÓNSSON H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. The Journal of Chemical Physics, 2000, 113(22):9978-9985.
DOI URL |
| [52] |
GEIM A K, NOVOSELOV K S. The rise of graphene. Nature Materials, 2007, 6(3):183-191.
DOI URL |
| [53] |
SONG E H, WEN Z, JIANG Q. CO catalytic oxidation on copper-embedded graphene. The Journal of Physical Chemistry C, 2011, 115(9):3678-3683.
DOI URL |
| [54] | SONG E H, YAN J M, LIAN J S, et al. External electric field catalyzed N2O decomposition on Mn-embedded graphene. The Journal of Physical Chemistry C, 2012, 116(38):20342-20348. |
| [55] | MAITARAD P, JUNKAEW A, PROMARAK V, et al. Complete catalytic cycle of NO decomposition on a silicon-doped nitrogen- coordinated graphene: mechanistic insight from a DFT study. Applied Surface Science, 2020, 508:145255. |
| [56] | LÜ Y A, ZHUANG G L, WANG J G, et al. Enhanced role of Al or Ga-doped graphene on the adsorption and dissociation of N2O under electric field. Phys. Chem. Chem. Phys., 2011, 13(27):12472-12477. |
| [1] | JIANG Yiyi, SHEN Min, SONG Banxia, LI Nan, DING Xianghuan, GUO Leyi, MA Guoqiang. Effect of Dual-functional Electrolyte Additive on High Temperature and High Voltage Performance of Li-ion Battery [J]. Journal of Inorganic Materials, 2022, 37(7): 710-716. |
| [2] | SUN Lian, GU Quanchao, YANG Yaping, WANG Honglei, YU Jinshan, ZHOU Xingui. Two-dimensional Transition Metal Dichalcogenides for Electrocatalytic Oxygen Reduction Reaction [J]. Journal of Inorganic Materials, 2022, 37(7): 697-709. |
| [3] | WANG Peng, JIN Zunlong, CHEN Ningguang, LIU Yonghao. Theoretical Investigation of Mo Doped α-MnO2 Electrocatalytic Oxygen Evolution Reaction [J]. Journal of Inorganic Materials, 2022, 37(5): 541-546. |
| [4] | WU Jing, YU Libing, LIU Shuaishuai, HUANG Qiuyan, JIANG Shanshan, ANTON Matveev, WANG Lianli, SONG Erhong, XIAO Beibei. NiN4/Cr Embedded Graphene for Electrochemical Nitrogen Fixation [J]. Journal of Inorganic Materials, 2022, 37(10): 1141-1148. |
| [5] | WANG Haoxuan, LIU Qiaomu, WANG Yiguang. Research Progress of High Entropy Transition Metal Carbide Ceramics [J]. Journal of Inorganic Materials, 2021, 36(4): 355-364. |
| [6] | ZHANG Ruihong, WEI Xin, LU Zhanhui, AI Yuejie. Training Model for Predicting Adsorption Energy of Metal Ions Based on Machine Learning [J]. Journal of Inorganic Materials, 2021, 36(11): 1178-1184. |
| [7] | HUANG Chong,ZHAO Wei,WANG Dong,BU Kejun,WANG Sishun,HUANG Fuqiang. Synthesis, Crystal Structure, and Electrical Conductivity of Pd-intercalated NbSe2 [J]. Journal of Inorganic Materials, 2020, 35(4): 505-510. |
| [8] | ZHOU Zihang, WANG Qun, GE Xiang, LI Zhaoyang. Strontium Doped Hydroxyapatite Nanoparticles: Synthesis, Characterization and Simulation [J]. Journal of Inorganic Materials, 2020, 35(11): 1283-1289. |
| [9] | QI Xin-Xin, SONG Guang-Ping, YIN Wei-Long, WANG Ming-Fu, HE Xiao-Dong, ZHENG Yong-Ting, WANG Rong-Guo, BAI Yue-Lei. Analysis on Phase Stability and Mechanical Property of Newly-discovered Ternary Layered Boride Cr4AlB4 [J]. Journal of Inorganic Materials, 2020, 35(1): 53-60. |
| [10] | ZOU Can-Hui, LONG Ying, ZHENG Xin, ZHANG Jin-Yang, LIN Hua-Tai. Preparation, Microstructure and Property of Ternary Transition Metal Boride Os1-xRuxB2 [J]. Journal of Inorganic Materials, 2018, 33(7): 787-792. |
| [11] | WANG Jun-Kai, ZHANG Yuan-Zhuo, LI Jun-Yi, ZHANG Hai-Jun, LI Fa-Liang, HAN Lei, SONG Shu-Peng. Low Temperature Catalytic Synthesis of β-SiC Powders via Microwave Heating [J]. Journal of Inorganic Materials, 2017, 32(7): 725-730. |
| [12] | TIAN Xiao-Dong, LI Xiao, YANG Tao, SONG Yan, LIU Zhan-Jun, GUO Quan-Gui. Recent Advances on Synthesis and Supercapacitor Application of Binary Metal Oxide [J]. Journal of Inorganic Materials, 2017, 32(5): 459-468. |
| [13] | CHEN Hai-Tao, HUANG Xue-Fei, HUANG Wei-Gang. Influence of N Doping on the Electronic Structure and Absorption Spectrum of Ca2SiO4: Eu2+ Phosphor [J]. Journal of Inorganic Materials, 2017, 32(4): 443-448. |
| [14] | ZHANG Chen-Le, ZHANG Pei-Xin, YUN Si-Ning, LI Yong-Liang, HE Ting-Shu. Recent Progress on Preparation of Transition Metal Compounds as Counter Electrodes for Dye-sensitized Solar Cells [J]. Journal of Inorganic Materials, 2016, 31(2): 113-122. |
| [15] | YANG Zhi-Huai, ZHANG Yun-Peng, ZHANG Mei-Guang, XU Qiang, ZHANG Ya-Ni, ZHANG Rong. The Electronic and Optical Properties of Tetrahedral Doped Co1-xRexCr2O4 (Re = Li, Na, K, Rb) Spinel [J]. Journal of Inorganic Materials, 2015, 30(8): 819-824. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||