Journal of Inorganic Materials ›› 2021, Vol. 36 ›› Issue (11): 1125-1136.DOI: 10.15541/jim20200683
Special Issue: 【虚拟专辑】电致变色与热致变色材料
• REVIEW • Next Articles
ZHAO Linyan1( ), LIU Yangsi1,2,3, XI Xiaoli1,2,4(), MA Liwen1,2, NIE Zuoren1,2,4
), LIU Yangsi1,2,3, XI Xiaoli1,2,4(), MA Liwen1,2, NIE Zuoren1,2,4
Received:2020-11-28
Revised:2021-04-26
Published:2021-11-20
Online:2021-06-01
Contact:
XI Xiaoli, professor. E-mail: xixiaoli@bjut.edu.cn
About author:ZHAO Linyan(1992-), female, PhD candidate. E-mail: zlyding@emails.bjut.edu.cn
Supported by:CLC Number:
ZHAO Linyan, LIU Yangsi, XI Xiaoli, MA Liwen, NIE Zuoren. First-principles Study on Nanoscale Tungsten Oxide: a Review[J]. Journal of Inorganic Materials, 2021, 36(11): 1125-1136.
| Type of tungsten oxide | Configuration | 3D Model |
|---|---|---|
| Cubic WO3 | $\text{pm\bar{3}m}\left( 221 \right)$ | |
| Hexagonal WO3 | $\text{p}6/\text{mmm}\left( 191 \right)$ | |
| Tetragonal WO3 | $\text{p}4/\text{ncc}\left( 130 \right)$ | |
| Orthorhombic WO3 | $\text{pbcn}\left( 60 \right)$ | |
| Monoclinic WO3 | \[\text{p}{{2}_{1}}\text{/c}\left( 14 \right)\] | |
| Triclinic WO3 | $\text{p}1\left( 1 \right)$ | |
| Orthorhombic WO2 | $\text{pnma}\left( 62 \right)$ | |
| Monoclinic WO3-x | $\text{p}2/\text{m}\left( 10 \right)$ | |
Table 1 Tungsten oxides with different crystal structures, space groups and 3D models (O and W atoms are represented by red and blue balls, respectively)
| Type of tungsten oxide | Configuration | 3D Model |
|---|---|---|
| Cubic WO3 | $\text{pm\bar{3}m}\left( 221 \right)$ | |
| Hexagonal WO3 | $\text{p}6/\text{mmm}\left( 191 \right)$ | |
| Tetragonal WO3 | $\text{p}4/\text{ncc}\left( 130 \right)$ | |
| Orthorhombic WO3 | $\text{pbcn}\left( 60 \right)$ | |
| Monoclinic WO3 | \[\text{p}{{2}_{1}}\text{/c}\left( 14 \right)\] | |
| Triclinic WO3 | $\text{p}1\left( 1 \right)$ | |
| Orthorhombic WO2 | $\text{pnma}\left( 62 \right)$ | |
| Monoclinic WO3-x | $\text{p}2/\text{m}\left( 10 \right)$ | |
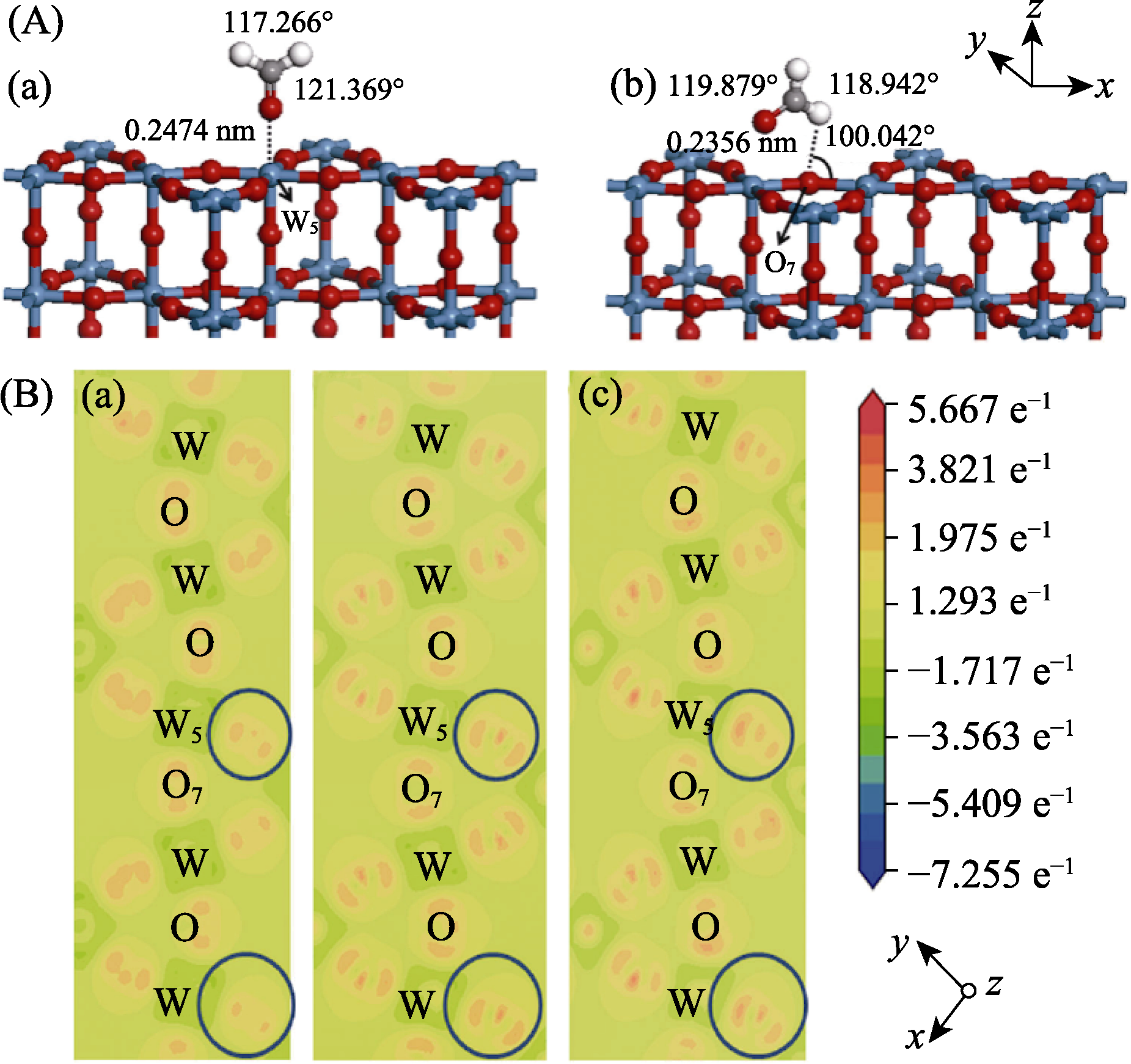
Fig. 1 (A) Optimized adsorption structures of HCHO with red, white and black balls representing O, H and C, respectively, on W5 (HCHO-W5 configuration) (a) and O7 (HCOH-O7 configuration) (b) sites of WO-terminated h-WO3 (001) surface; (B) Calculated electron density difference of the clean (001) surface (a), HCHO-absorbed on (001) surface for HCHO-W5 (b) and HCOH-O7 (c) configurations[59]
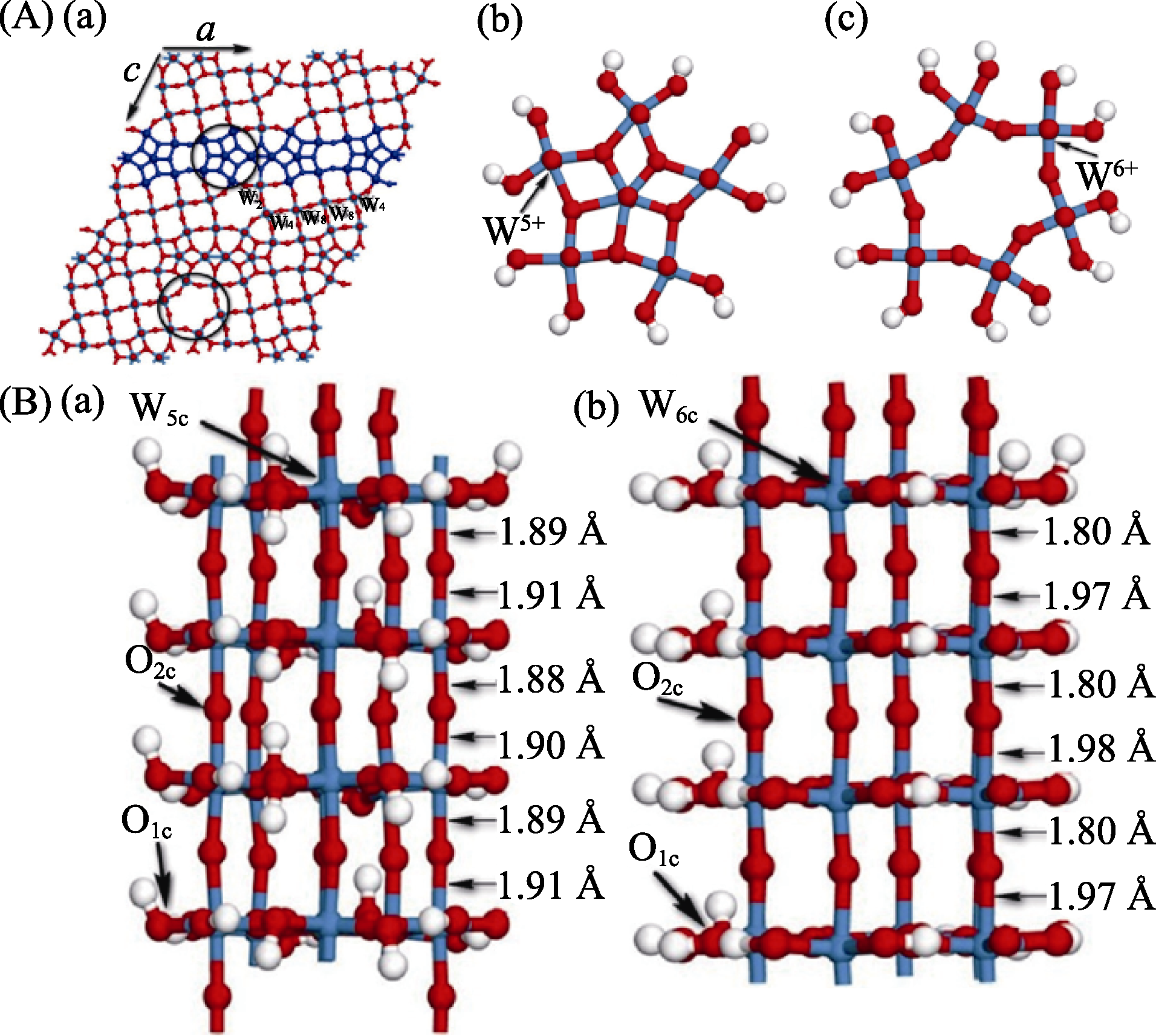
Fig. 2 (A) Monoclinic structure (a) of W18O49 nanowires supercell model and its top views of NW1(b) and NW2(c), where NW1 and NW2 include largely cations W5+ and cations W6+, respectively; (B) Optimized models for NW1 (a) and NW2 (b), of W18O49 (010) nanowires[24, 61-62]. O, W and H atoms are represented by red, blue and white balls, respectively (1 Å=0.1 nm)
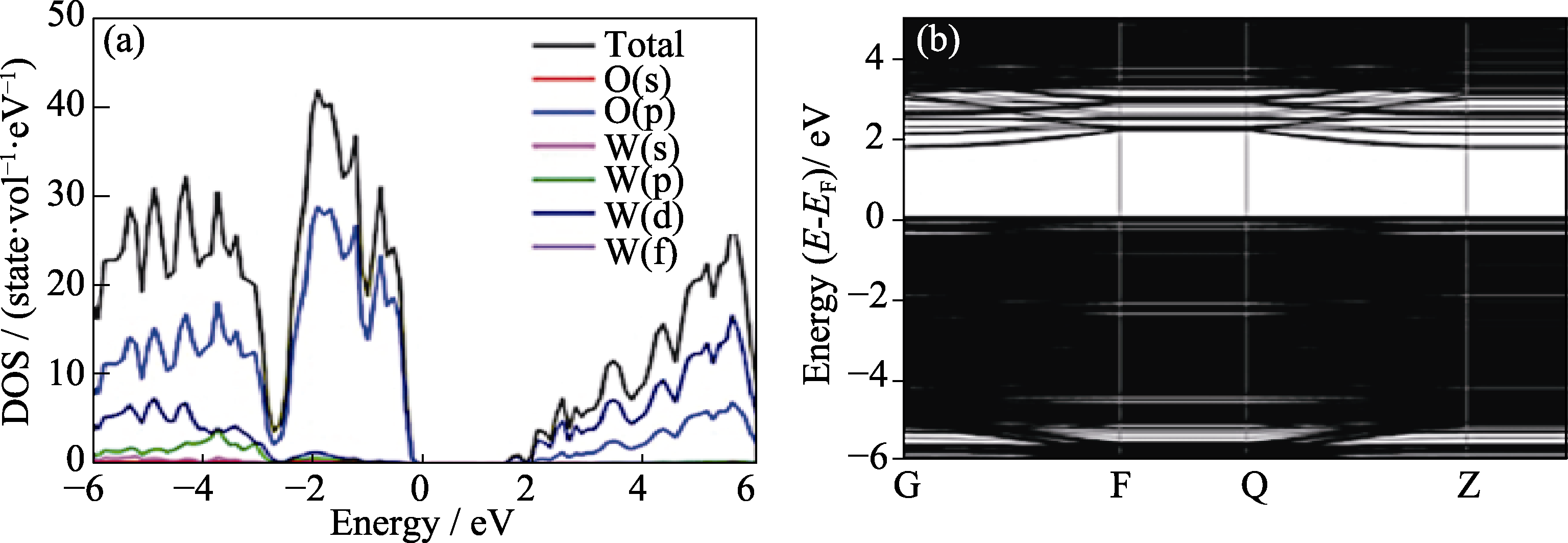
Fig. 3 (a) Density of states and projected density of states of bulk WO3 without oxygen vacancy, and (b) structure of WO3(002) with one oxygen vacancy[28] Colorful images showing on website
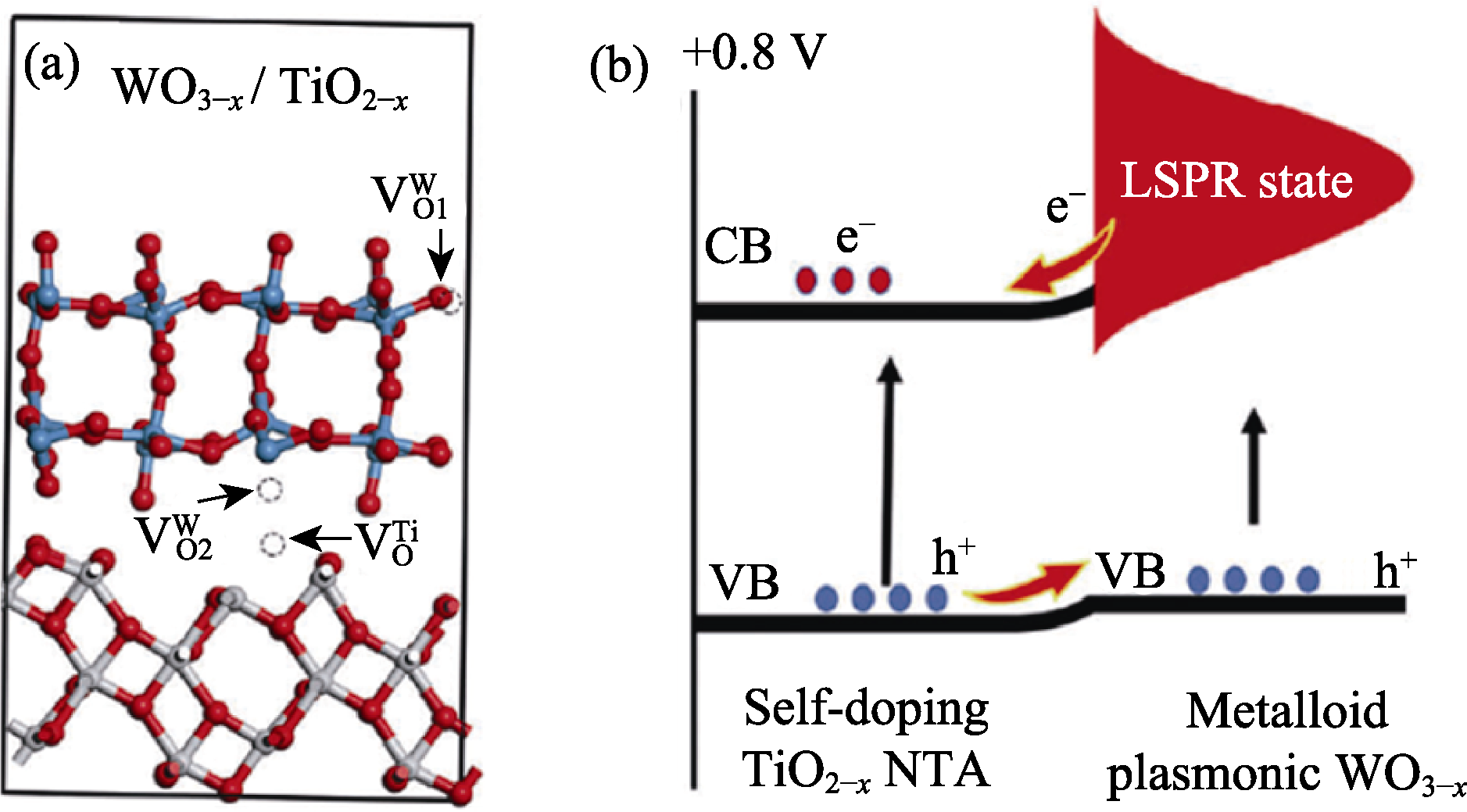
Fig. 4 (a) Geometrical optimized equilibrium configuration of WO3-x/TiO2-x with red, blue and white balls representing O, W and Ti, respectively, and (b) schematic diagram of the self-doping Ti3+, localized surface plasmon resonance (LSPR), and charge transfer in WO3-x/TiO2-x[72]
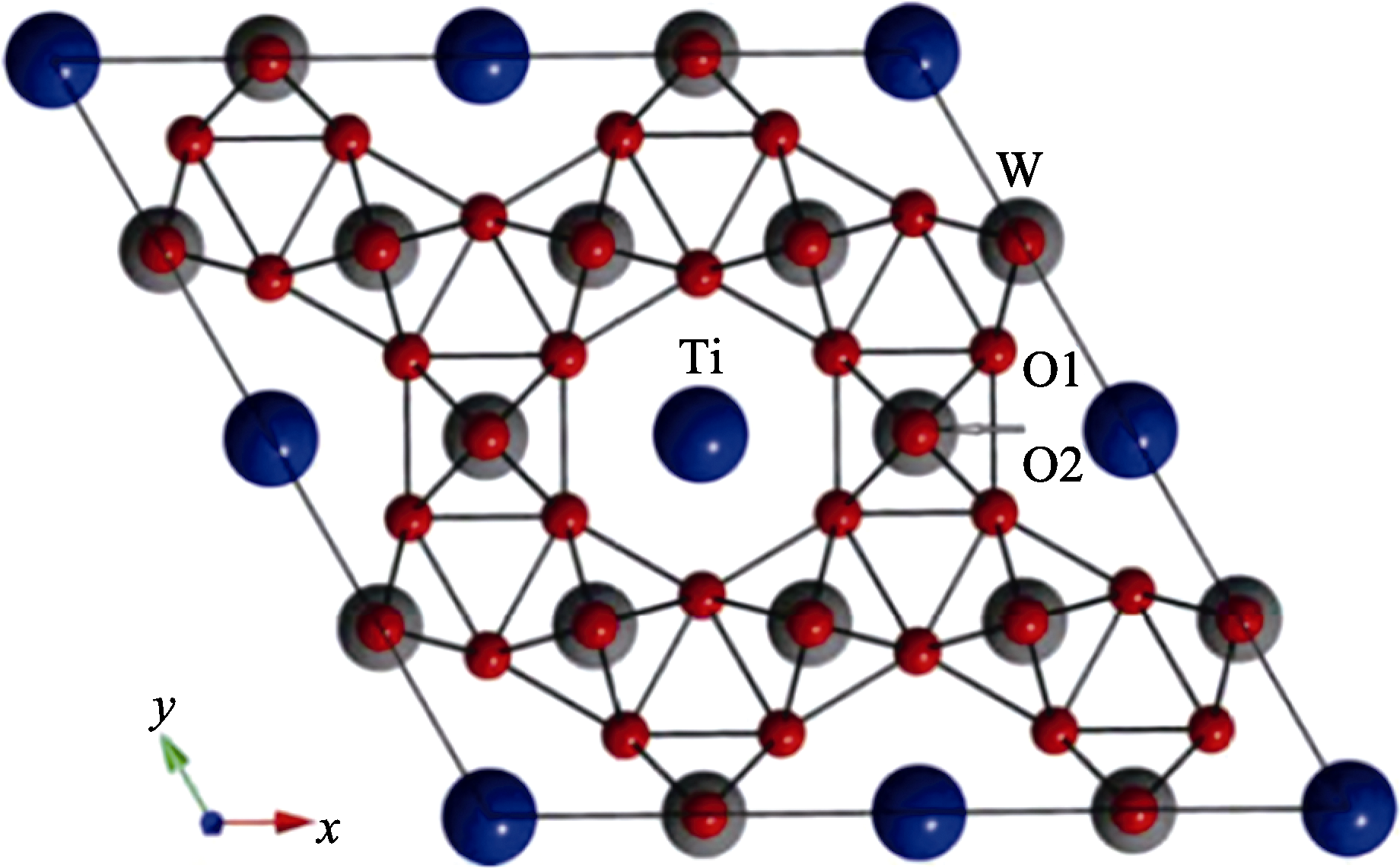
Fig. 5 Top view of the supercell of Ti-doped h-WO3[29]
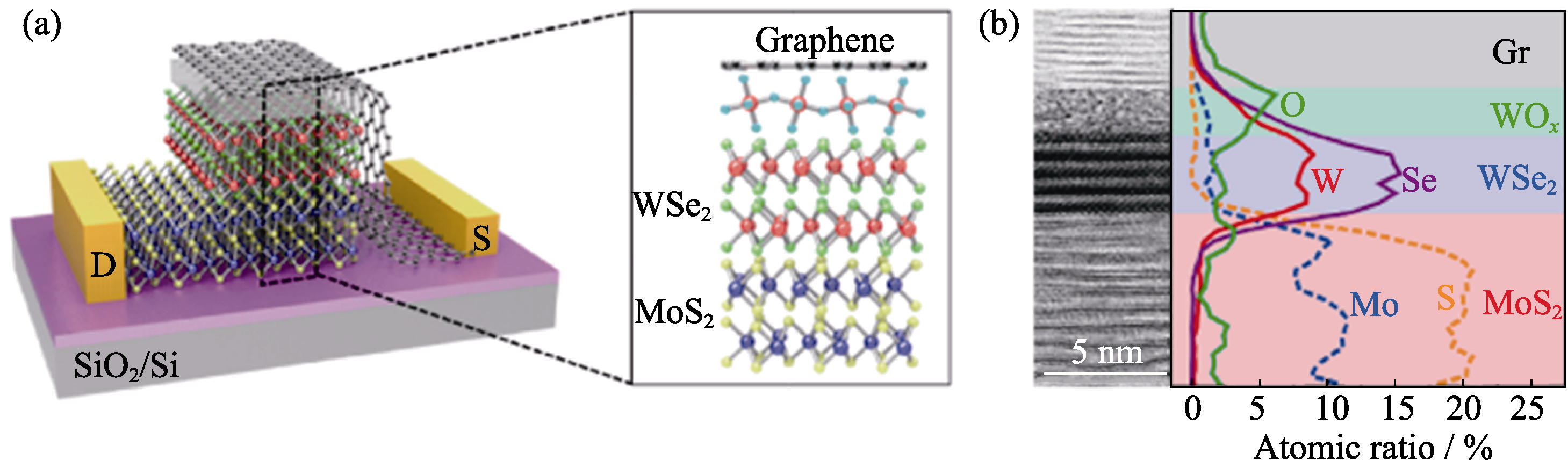
Fig. 6 Monolithically band-engineered WSe2-MoS2 p-n heterojunction[82] (a) Schematic illustration of the vertical WSe2-MoS2 device with the WOx layer, where pink, blue, green, purple and yellow balls represent W, O, Se, Mo and S, respectively; (b) Cross-sectional HR-TEM image and EDS elemental line profiles across the WOx/WSe2/MoS2 heterointerfaces
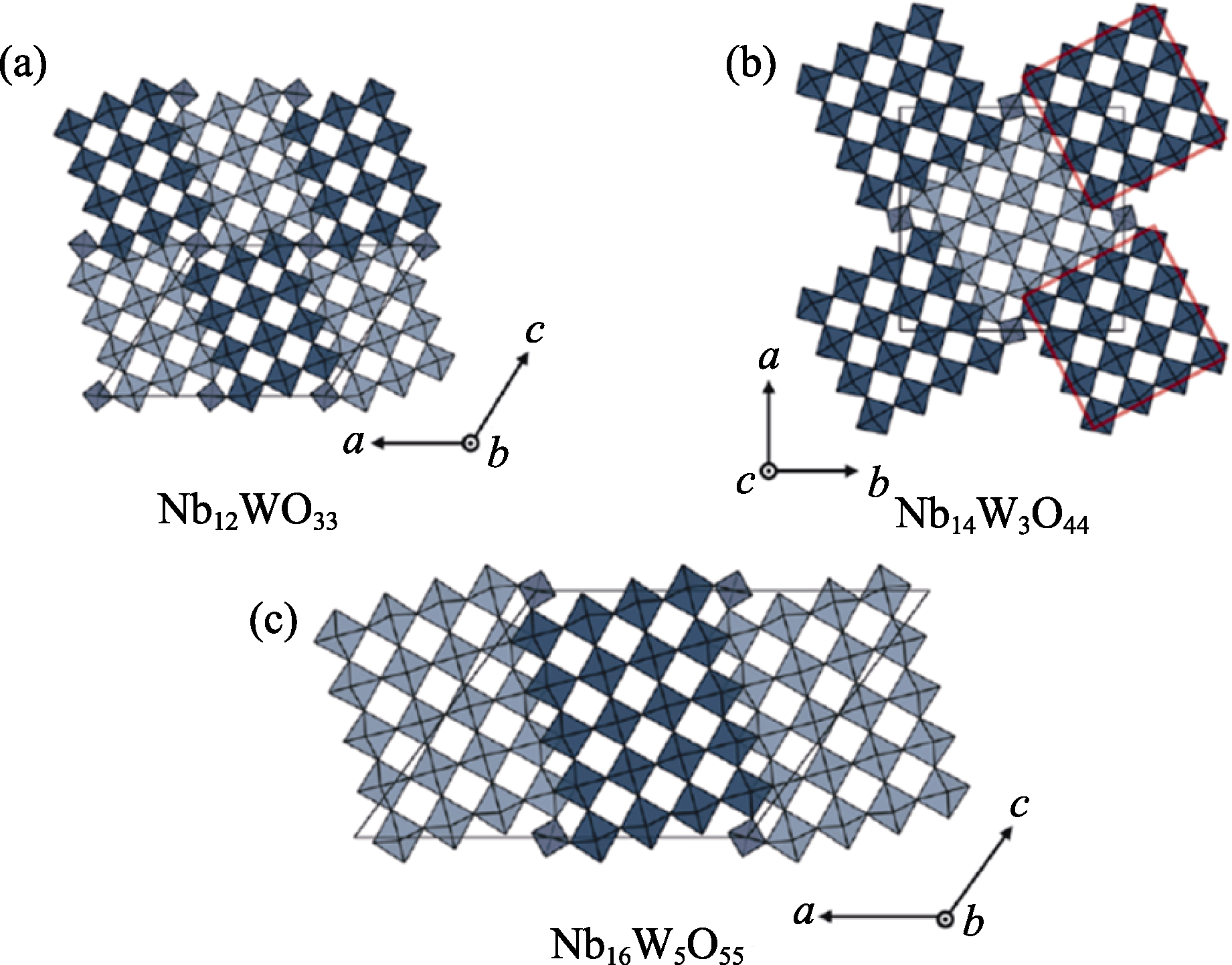
Fig. 7 Crystal structures of Wadsley-Roth phases[85] (a) Nb12WO33 (space group C2); (b) Nb14W3O44 (I4/m); (c) Nb16W5O55 (C2)
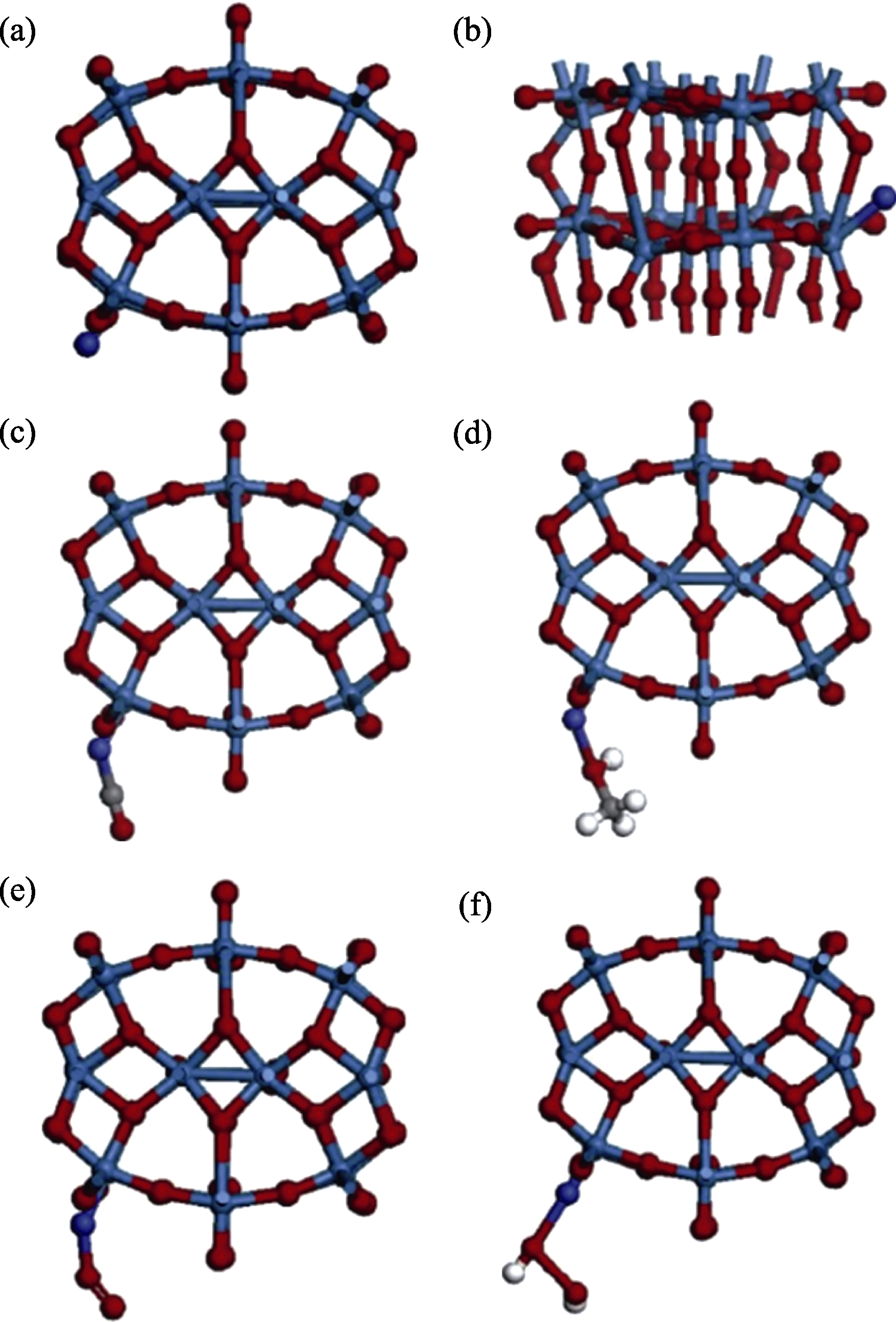
Fig. 8 Optimized sadsorption model of different gas molecules on W18O49 with blue, purple, red, gray and white balls represent W, Co, O, C and H, respectively[86] (a, b) Adsorbed cobalt atom on the tungsten atom of NW (NW-Co); (c) Carbon monoxide molecule adsorbed on the NW-Co; (d) Methanol molecule adsorbed on the NW-Co; (e) Oxygen molecule adsorbed on the NW-Co; (f) Hydrogen peroxide molecule adsorbed on the NW-Co
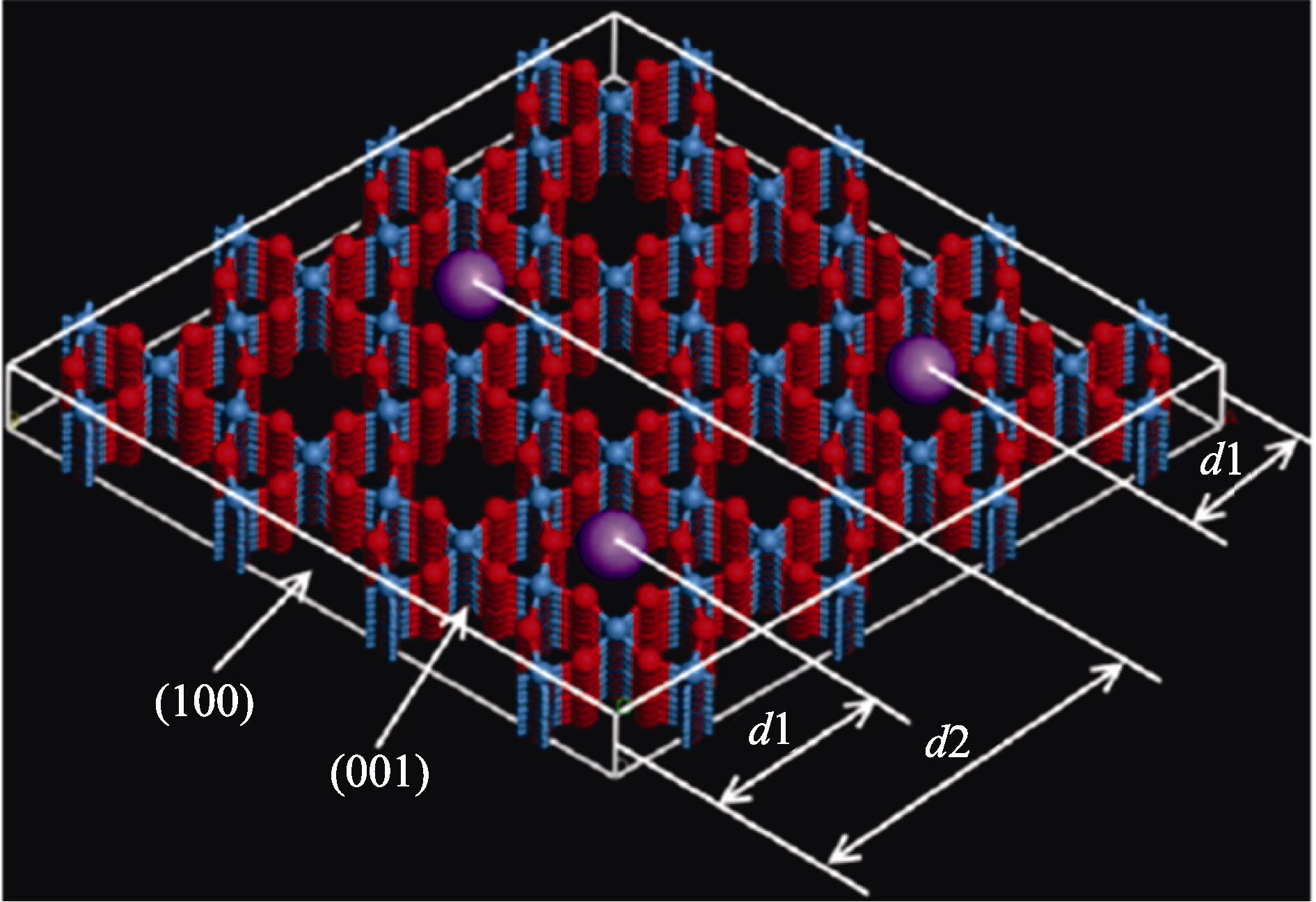
Fig. 9 Various intercalating sites corresponding to different distances to the h-WO3(100) surface with blue, red and purple balls representing W, O and cations, respectively[90]
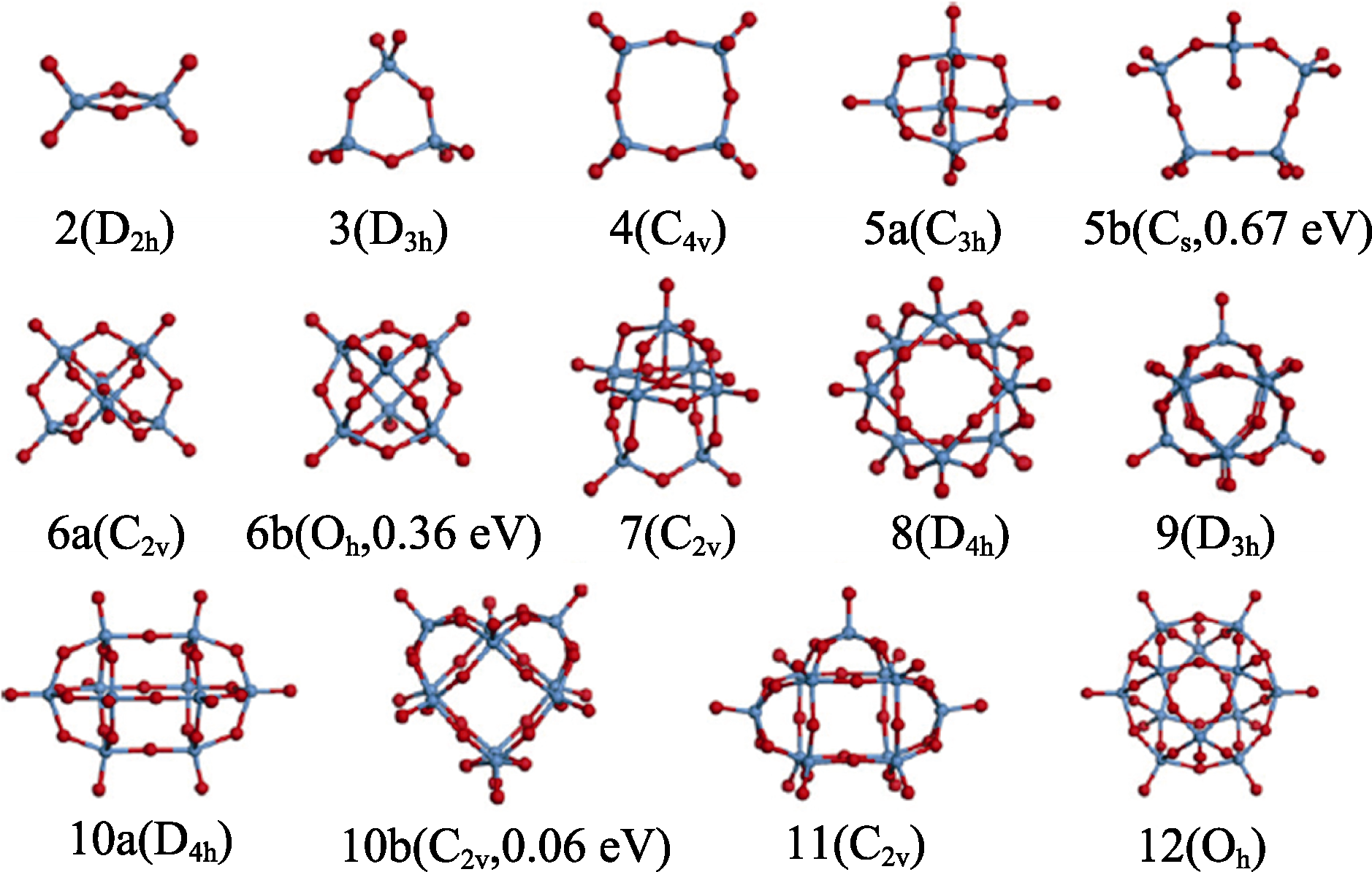
Fig. 10 Lowest-energy structures of (WO3)n clusters (n=2-12) and several metastable isomers (labeled as 5b, 6b, 10b) with blue and red balls representing W and O, respectively[95]
| [1] |
YIN X T, LV P, LI J, et al. Nanostructured tungsten trioxide prepared at various growth temperatures for sensing applications. Journal of Alloys and Compounds, 2020, 825:154105.
DOI URL |
| [2] |
NANDIYANTO A B D, OKTIANI R, RAGADHITA R, et al. Amorphous content on the photocatalytic performance of micrometer-sized tungsten trioxide particles. Arabian Journal of Chemistry, 2020, 13(1):2912-2924.
DOI URL |
| [3] |
HE X, WANG X Y, SUN B N, et al. Synthesis of three- dimensional hierarchical furball-like tungsten trioxide microspheres for high performance supercapacitor electrodes. RSC Advances, 2020, 10(23):13437-13441.
DOI URL |
| [4] |
HAI G J, HUANG J F, CAO L Y, et al. Influence of oxygen deficiency on the synthesis of tungsten oxide and the photocatalytic activity for the removal of organic dye. Journal of Alloys and Compounds, 2017, 690:239-248.
DOI URL |
| [5] |
LIU X F, ZHOU H, PEI S Z, et al. Oxygen-deficient WO3-x nanoplate array film photoanode for efficient photoelectrocatalytic water decontamination. Chemical Engineering Journal, 2020, 381:122740.
DOI URL |
| [6] | 赵林艳, 席晓丽, 樊佑书, 等. 纳米氧化钨的水热/溶剂热法制备及应用的综述. 材料导报, 2019, 33(19):3203-3209. |
| [7] |
QUAN H Q, GAO Y F, WANG W Z. Tungsten oxide-based visible light-driven photocatalysts: crystal and electronic structures and strategies for photocatalytic efficiency enhancement. Inorganic Chemistry Frontiers, 2020, 7(4):817-838.
DOI URL |
| [8] | PERSSON K. Materials project. https://materialsproject.org/ . |
| [9] |
DEB S K. Opportunities and challenges in science and technology of WO3 for electrochromic and related applications. Solar Energy Materials and Solar Cells, 2008, 92(2):245-258.
DOI URL |
| [10] |
DING Y, YANG I S, LI Z Q, et al. Nanoporous TiO2 spheres with tailored textural properties: controllable synthesis, formation mechanism, and photochemical applications. Progress in Materials Science, 2020, 109:100620.
DOI URL |
| [11] |
DONG P Y, HOU G H, XI X U, et al. WO3-based photocatalysts: morphology control, activity enhancement and multifunctional applications. Environmental Science-Nano, 2017, 4(3):539-557.
DOI URL |
| [12] |
HAN L F, CHEN J L, ZHANG Y H, et al. Facile synthesis of hierarchical carpet-like WO3 microflowers for high NO2 gas sensing performance. Materials Letters, 2018, 210:8-11.
DOI URL |
| [13] |
LI Y S, TANG Z L, ZHANG J Y, et al. Fabrication of vertical orthorhombic/hexagonal tungsten oxide phase junction with high photocatalytic performance. Applied Catalysis B-Environmental, 2017, 207:207-217.
DOI URL |
| [14] |
HUNGE Y M, YADAV A A, MAHADIK M A, et al. A highly efficient visible-light responsive sprayed WO3/FTO photoanode for photoelectrocatalytic degradation of brilliant blue. Journal of the Taiwan Institute of Chemical Engineers, 2018, 85:273-281.
DOI URL |
| [15] | KARADENIZ S M, TATAR D, ERTUGRUL M, et al. Structural, optical and electrochromic properties of WO3 thin films prepared by chemical spray pyrolysis versus spin coating technique. Spectroscopy and Spectral Analysis, 2018, 38(9):2982-2988. |
| [16] |
INAMDAR A I, CHAVAN H S, AHMED A A, et al. Nanograin tungsten oxide with excess oxygen as a highly reversible anode material for high-performance Li-ion batteries. Materials Letters, 2018, 215:233-237.
DOI URL |
| [17] |
SHENG J P, ZHANG L, DENG L, et al. Fabrication of dopamine enveloped WO3-x quantum dots as single-NIR laser activated photonic nanodrug for synergistic photothermal/photodynamic therapy against cancer. Chemical Engineering Journal, 2020, 383:123071.
DOI URL |
| [18] |
ZHAO L Y, XI X L, LIU Y S, et al. Growth mechanism and visible-light-driven photocatalysis of organic solvent dependent WO3 and nonstoichiometric WO3-x nanostructures. Journal of the Taiwan Institute of Chemical Engineers, 2020, 115:339-347.
DOI URL |
| [19] |
ZHAO L Y, XI X L, LIU Y S, et al. Facile synthesis of WO3 micro/nanostructures by paper-assisted calcination for visible- light-driven photocatalysis. Chemical Physics, 2020, 528:110515.
DOI URL |
| [20] |
FAN Y S, XI X L, LIU Y S, et al. Growth mechanism of immobilized WO3 nanostructures in different solvents and their visible-light photocatalytic performance. Journal of Physics and Chemistry of Solids, 2020, 140:109380.
DOI URL |
| [21] |
MANTHIRAM K, ALIVISATOS A P. Tunable localized surface plasmon resonances in tungsten oxide nanocrystals. Journal of the American Chemical Society, 2012, 134(9):3995-3998.
DOI URL |
| [22] |
KIMURA Y, IBANO K, UEHATA K, et al. Improved hydrogen gas sensing performance of WO3 films with fibrous nanostructured surface. Applied Surface Science, 2020, 532:147274.
DOI URL |
| [23] |
LI D, HUANG W Q, XIE Z, et al. Mechanism of enhanced photocatalytic activities on tungsten trioxide doped with sulfur: dopant-type effects. Modern Physics Letters B, 2016, 30(27):1650340.
DOI URL |
| [24] |
QIN Y X, LIU M, YE Z H. A DFT study on WO3 nanowires with different orientations for NO2 sensing application. Journal of Molecular Structure, 2014, 1076:546-553.
DOI URL |
| [25] |
CHEN Z J, CAO J X, YANG L W, et al. The unique photocatalysis properties of a 2D vertical MoO2 /WO2 heterostructure: a first- principles study. Journal of Physics D-Applied Physics, 2018, 51(26):265106.
DOI URL |
| [26] | 张秋杰, 高占忠, 原玉, 等. 第一性原理研究荷电状态对Pd13团簇催化分解NO性能的影响. 原子与分子物理学报, 2016, 33(03):438-442. |
| [27] |
JIA Q Q, JI H M, BAI X. Selective sensing property of triclinic WO3 nanosheets towards ultra-low concentration of acetone. Journal of Materials Science-Materials in Electronics, 2019, 30(8):7824-7833.
DOI URL |
| [28] |
MA Y L, FENG B, LANG J Y, et al. Synthesis of semimetallic tungsten trioxide for infrared light photoelectrocatalytic water splitting. Journal of Physical Chemistry C, 2019, 123(42):25833-25843.
DOI URL |
| [29] | 秦京运, 舒群威, 袁艺, 等. Ti0.33WO3电子结构和太阳辐射屏蔽性能第一性原理研究. 物理学报, 2020, 69(4):217-223. |
| [30] | DIRAC P A M. The Principles of Quantum Mechanics. Oxford: Clarendon Press, 1958: 1-22. |
| [31] | 曾瑾言. 量子力学. 北京, 科学出版社, 2000: 1-24. |
| [32] | BORN M, HUANG K. Dynamical Theory of Crystal Lattices. Oxford: Oxford University Press, 1958: 104-113. |
| [33] |
SLATER J C. Magnetic effects and the Hartree-Fock equation. Physical Review, 1951, 82(4):538-541.
DOI URL |
| [34] | 张跃, 谷景华, 商家香, 等. 计算材料学基础. 北京: 北京航天航空大学出版社, 2007: 83-135. |
| [35] | 徐光宪, 黎乐民, 王德民. 量子化学:基本原理和从头计算法. 北京: 科学出版社, 2007: 65-97. |
| [36] |
KOCH W, HOLTHAUSEN M C. A chemist's guide to density functional theory. Zeitschrift für Physik B Condensed Matter, 2001, 78(2):317-323.
DOI URL |
| [37] | KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects. Physical Review, 1965, 140:1133-1138. |
| [38] |
THOMAS H. The calculation of atomic fields. Proceedings of the Cambridge Philosophical Society, 1927, 23:542-548.
DOI URL |
| [39] |
KOHN W. Nobel lecture: electronic structure of matter-wave functions and density functionals. Reviews of Modern Physics, 1999, 71(5):1253-1266.
DOI URL |
| [40] | FERMI E. Un metodo statistico per la determinazione di alcune Priorieta dell atome. Rend. Accad. Naz. Lincei, 1927, 6:602. |
| [41] | DIRAC P A M. Note on exchange phenomena in the thomas-fermi atom. Proceedings of the Cambridge Philosophical Royal Society, 1930, 26:376. |
| [42] | SLATER J C. A simplification of the hartree-fock method. Self-Consistent Fields in Atoms, 1975, 81(3):215-230. |
| [43] |
PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, molecules, solids, and surfaces-applications of the generalized gradient approximation for exchange and correlation. Physical Review B, 1992, 46(11):6671-6687.
DOI URL |
| [44] | 谢希德, 陆栋. 固体能带理论. 上海: 复旦大学出版社, 1998: 66-69. |
| [45] |
VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Physical Review B, 1990, 41(11):7892-7895.
DOI URL |
| [46] |
BLÖCHL P E. Projector augmented-wave method. Physical Review B Condens Matter, 1994, 50(24):17953-17959.
DOI URL |
| [47] |
HAMANN D R, SCHLÜTER M, CHIANG C. Norm-conserving pseudopotentials. Physical Review Letters, 1979, 43(20):1494-1497.
DOI URL |
| [48] | 北京创腾科技有限公司. BIOVIA materials studio.[EB/OL]. [2021-04-26] https://www.neotrident.com/index.php/product/proinfo/29.html . |
| [49] | 北京宏剑公司. Vienna ab-initio simulation package. [EB/OL]. [2021-04-26] http://www.hongcam.com.cn/index.php/product/materials/vasp . |
| [50] | WEB V. Psi-k-Ab initio. [EB/OL]. [2021-04-26] http://psi-k.net/software/ . |
| [51] | KAMINSKY W. WinXMorph. [EB/OL]. [2021-04-26] http://cad4.cpac.washington.edu/WinXMorphHome/WinXMorph.htm#opennewwindow . |
| [52] | MOMMA K. Visualization for electronic and structural analysis. [EB/OL]. (2021-01-09) [2021-04-26] http://www.jp-minerals.org/vesta/en/ . |
| [53] |
MAHAJAN S, JAGTAP S. Metal-oxide semiconductors for carbon monoxide (CO) gas sensing: a review. Applied Materials Today, 2020, 18:100483.
DOI URL |
| [54] |
ZEB S, PENG X J, YUAN G Z, et al. Controllable synthesis of ultrathin WO3 nanotubes and nanowires with excellent gas sensing performance. Sensors and Actuators B-Chemical, 2020, 305:127435.
DOI URL |
| [55] |
LIU D, REN X W, LI Y S, et al. Nanowires-assembled WO3 nanomesh for fast detection of ppb-level NO2 at low temperature. Journal of Advanced Ceramics, 2020, 9(2):17-26.
DOI URL |
| [56] |
OISON V, SAADI L, LAMBERT-MAURIAT C, et al. Mechanism of CO and O3 sensing on WO3 surfaces: first principle study. Sensors and Actuators B-Chemical, 2011, 160(1):505-510.
DOI URL |
| [57] |
ZHAO L H, TIAN F H, WANG X B, et al. Mechanism of CO adsorption on hexagonal WO3(001) surface for gas sensing: a DFT study. Computational Materials Science, 2013, 79:691-697.
DOI URL |
| [58] |
JIN H, ZHOU H, ZHANG Y F. Insight into the mechanism of CO oxidation on WO3(001) surfaces for gas sensing: a DFT study. Sensors, 2017, 17(8):1898.
DOI URL |
| [59] | TANG B L, JIANG G H, CHEN W X, et al. First-principles study on hexagonal WO3 for HCHO gas sensing application. Acta Metallurgica Sinica-English Letters, 2015, 28(6):772-780. |
| [60] | HAN X, YIN X H. Density functional theory study of the NO2-sensing mechanism on a WO3(001) surface: the role of surface oxygen vacancies in the formation of NO and NO3. Molecular Physics, 2016, 114(24):3546-3555. |
| [61] | QIN Y X, LIU M, HUA D Y. First-principles study of the electronic structure and NO2-sensing properties of Ti-doped W18O49 nanowire. Acta Physica Sinica, 2014, 63(20):207101. |
| [62] | QIN Y X, YE Z H. DFT study on interaction of NO2 with the vacancy-defected WO3 nanowires for gas-sensing. Sensors and Actuators B-Chemical, 2016, 222:499-507. |
| [63] | BAI S L, ZHANG K W, WANG L S, et al. Synthesis mechanism and gas-sensing application of nanosheet-assembled tungsten oxide microspheres. Journal of Materials Chemistry A, 2014, 2(21):7927-7934. |
| [64] | YAKOVKIN I N, GUTOWSKI M. Driving force for the WO3(001) surface relaxation. Surface Science, 2007, 601(6):1481-1488. |
| [65] | 张克伟. 气敏和光催化导向的氧化钨纳米结构设计与改性. 北京: 北京化工大学博士学位论文, 2014. |
| [66] | YANG H H, SUN H G, LI Q T, et al. Structural, electronic, optical and lattice dynamic properties of the different WO3 phases: first-principle calculation. Vacuum, 2019, 164:411-420. |
| [67] | 杨欢欢. 第一性原理研究WO3低指数表面上H2O分子吸附和分解的微观机制. 济南: 山东大学硕士学位论文, 2019. |
| [68] | ZHENG T T, SANG W, HE Z H, et al. Conductive tungsten oxide nanosheets for highly efficient hydrogen evolution. Nano Letters, 2017, 17(12):7968-7973. |
| [69] | 桑炜. 纳米晶可控合成以及催化性能的调控. 合肥: 中国科学技术大学博士学位论文, 2018. |
| [70] | WANG F G, DI VALENTIN C, PACCHIONI G. Doping of WO3 for photocatalytic water splitting: hints from density functional theory. Journal of Physical Chemistry C, 2012, 116(16):8901-8909. |
| [71] | ZHANG T, ZHU Z L, CHEN H N, et al. Iron-doping-enhanced photoelectrochemical water splitting performance of nanostructured WO3: a combined experimental and theoretical study. Nanoscale, 2015, 7(33):2933-2940. |
| [72] | HUANG W C, WANG J X, BIAN L, et al. Oxygen vacancy induces self-doping effect and metalloid LSPR in non-stoichiometric tungsten suboxide synergistically contributing to the enhanced photoelectrocatalytic performance of WO3-x/TiO2-x heterojunction. Physical Chemistry Chemical Physics, 2018, 20(25):17268-17278. |
| [73] | ZHANG N, LI X Y, LIU Y F, et al. Defective tungsten oxide hydrate nanosheets for boosting aerobic coupling of amines: synergistic catalysis by oxygen vacancies and bronsted acid sites. Small, 2017, 13(31):1701354. |
| [74] | ZHANG N, JALIL A, WU D X, et al. Refining defect states in W18O49 by Mo doping: a strategy for tuning N2 activation towards solar-driven nitrogen fixation. Journal of the American Chemical Society, 2018, 140(30):9434-9443. |
| [75] | ZHANG N, LONG R, GAO C, et al. Recent progress on advanced design for photoelectrochemical reduction of CO2 to fuels. Science China-Materials, 2018, 61(6):771-805. |
| [76] | LI M Q, ZHANG N, LONG R, et al. PdPt alloy nanocatalysts supported on TiO2: maneuvering metal-hydrogen interactions for light-driven and water-donating selective alkyne semihydrogenation. Small, 2017, 13(23):1604173. |
| [77] | WANG Z, WANG X Y, CONG S, et al. Fusing electrochromic technology with other advanced technologies: a new roadmap for future development. Materials Science & Engineering R-Reports, 2020, 140:100524. |
| [78] | YAO Y J, ZHAO Q, WEI W, et al. WO3 quantum-dots electrochromism. Nano Energy, 2020, 68:104350. |
| [79] | LIN H, ZHOU F, LIU C P, et al. Non-grotthuss proton diffusion mechanism in tungsten oxide dihydrate from first-principles calculations. Journal of Materials Chemistry A, 2014, 2(31):12280-12288. |
| [80] | HJELM A, GRANQVIST C G, WILLS J M. Electronic structure and optical properties of WO3, LiWO3, NaWO3, and HWO3. Physical Review B, 1996, 54(4):2436-2445. |
| [81] | WISEMAN P J, DICKENS P G. Neutron-diffraction studies of cubic tungsten bronzes. Journal of Solid State Chemistry, 1976, 17(1/2):91-100. |
| [82] | YANG S, CHA J, KIM J C, et al. Monolithic interface contact engineering to boost optoelectronic performances of 2D semiconductor photovoltaic heterojunctions. Nano Letters, 2020, 20(4):2443-2451. |
| [83] | 方城, 汪洪, 施思齐. 氧化钨电致变色性能的研究进展. 物理学报, 2016, 65(16):168201. |
| [84] | 高占忠. 锂离子电池负极材料WO3的第一性原理研究. 成都: 电子科技大学硕士学位论文, 2017. |
| [85] | KOCER C P, GRIFFITH K J, GREY C P, et al. Cation disorder and lithium insertion mechanism of Wadsley-Roth crystallographic shear phases from first principles. Journal of the American Chemical Society, 2019, 141(38):15121-15134. |
| [86] | KARIM N A, KAMARUDIN S K, SHYUAN L K, et al. Study on the electronic properties and molecule adsorption of W18O49 nanowires as a catalyst support in the cathodes of direct methanol fuel cells. Journal of Power Sources, 2015, 288:461-472. |
| [87] | ZHANG Z F, CHEN J L, LI H B, et al. Vapor-solid nanotube growth via sidewall epitaxy in an environmental transmission electron microscope. Crystal Growth & Design, 2017, 17(1):11-15. |
| [88] | ZHANG Z F, WANG Y, LI H B, et al. Atomic-scale observation of vapor-solid nanowire growth via oscillatory mass transport. ACS Nano, 2016, 10(1):763-769. |
| [89] | ZHANG Z F, SHENG L P, CHEN L, et al. Atomic-scale observation of pressure-dependent reduction dynamics of W18O49 nanowires using environmental TEM. Physical Chemistry Chemical Physics, 2017, 19(25):16307-16311. |
| [90] | CHEN L, LAM S, ZENG Q H, et al. Effect of cation intercalation on the growth of hexagonal WO3 nanorods. Journal of Physical Chemistry C, 2012, 116(21):11722-11727. |
| [91] | JIANG S, CHEKINI M, QU Z B, et al. Chiral ceramic nanoparticles and peptide catalysis. Journal of the American Chemical Society, 2017, 139(39):13701-13712. |
| [92] | GU L J, MA C L, ZHANG X H, et al. Populating surface-trapped electrons towards SERS enhancement of W18O49 nanowires. Chemical Communications, 2018, 54(49):6332-6335. |
| [93] | MEHMOOD F, PACHTER R, MURPHY N R, et al. Effect of oxygen vacancies on the electronic and optical properties of tungsten oxide from first principles calculations. Journal of Applied Physics, 2016, 120(23):233105. |
| [94] | MIGAS D B, SHAPOSHNIKOV V L, RODIN V N, et al. Tungsten oxides. I. Effects of oxygen vacancies and doping on electronic and optical properties of different phases of WO3. Journal of Applied Physics, 2010, 108(9):093713. |
| [95] | SAI L W, TANG L L, HUANG X M, et al. Lowest-energy structures of (WO3)n(2≤n≤12) clusters from first-principles global search. Chemical Physics Letters, 2012, 544:7-12. |
| [96] | HUANG X, ZHAI H J, LI J, et al. On the structure and chemical bonding of tri-tungsten oxide clusters W3On- and W3On (n=7-10): W3O8 as a potential molecular model for O-deficient defect sites in tungsten oxides. Journal of Physical Chemistry A, 2006, 110(1):85-92. |
| [97] | 邱克强, 王爱民, 张海峰, 等. 钨丝增强ZrAlNiCuSi块体非晶复合材料及其塑性行为. 金属学报, 2002(10):1091-1096. |
| [98] | JIANG P G, XIAO Y Y, LIU W J, et al. Hydrogen reduction characteristics of WO3 based on density functional theory. Results in Physics, 2019, 12:896-902. |
| [99] | LIU W J, JIANG P G, XIAO Y Y, et al. A study of the hydrogen adsorption mechanism of W18O49 using first-principles calculations. Computational Materials Science, 2018, 154:53-59. |
| [100] | 宋翰林, 姜平国, 刘文杰, 等. 氧化钨氢还原动力学的研究进展. 有色金属科学与工程, 2017, 8(5):64-69. |
| [101] | 姜平国, 汪正兵, 闫永播. 三氧化钨表面氢吸附机理的第一性原理研究. 物理学报, 2017, 66(8):294-303. |
| [1] | DING Ling, JIANG Rui, TANG Zilong, YANG Yunqiong. MXene: Nanoengineering and Application as Electrode Materials for Supercapacitors [J]. Journal of Inorganic Materials, 2023, 38(6): 619-633. |
| [2] | YANG Zhuo, LU Yong, ZHAO Qing, CHEN Jun. X-ray Diffraction Rietveld Refinement and Its Application in Cathode Materials for Lithium-ion Batteries [J]. Journal of Inorganic Materials, 2023, 38(6): 589-605. |
| [3] | CHEN Qiang, BAI Shuxin, YE Yicong. Highly Thermal Conductive Silicon Carbide Ceramics Matrix Composites for Thermal Management: a Review [J]. Journal of Inorganic Materials, 2023, 38(6): 634-646. |
| [4] | LIN Junliang, WANG Zhanjie. Research Progress on Ferroelectric Superlattices [J]. Journal of Inorganic Materials, 2023, 38(6): 606-618. |
| [5] | NIU Jiaxue, SUN Si, LIU Pengfei, ZHANG Xiaodong, MU Xiaoyu. Copper-based Nanozymes: Properties and Applications in Biomedicine [J]. Journal of Inorganic Materials, 2023, 38(5): 489-502. |
| [6] | YUAN Jingkun, XIONG Shufeng, CHEN Zhangwei. Research Trends and Challenges of Additive Manufacturing of Polymer-derived Ceramics [J]. Journal of Inorganic Materials, 2023, 38(5): 477-488. |
| [7] | DU Jianyu, GE Chen. Recent Progress in Optoelectronic Artificial Synapse Devices [J]. Journal of Inorganic Materials, 2023, 38(4): 378-386. |
| [8] | YANG Yang, CUI Hangyuan, ZHU Ying, WAN Changjin, WAN Qing. Research Progress of Flexible Neuromorphic Transistors [J]. Journal of Inorganic Materials, 2023, 38(4): 367-377. |
| [9] | YOU Junqi, LI Ce, YANG Dongliang, SUN Linfeng. Double Dielectric Layer Metal-oxide Memristor: Design and Applications [J]. Journal of Inorganic Materials, 2023, 38(4): 387-398. |
| [10] | QI Zhanguo, LIU Lei, WANG Shouzhi, WANG Guogong, YU Jiaoxian, WANG Zhongxin, DUAN Xiulan, XU Xiangang, ZHANG Lei. Progress in GaN Single Crystals: HVPE Growth and Doping [J]. Journal of Inorganic Materials, 2023, 38(3): 243-255. |
| [11] | ZHANG Chaoyi, TANG Huili, LI Xianke, WANG Qingguo, LUO Ping, WU Feng, ZHANG Chenbo, XUE Yanyan, XU Jun, HAN Jianfeng, LU Zhanwen. Research Progress of ScAlMgO4 Crystal: a Novel GaN and ZnO Substrate [J]. Journal of Inorganic Materials, 2023, 38(3): 228-242. |
| [12] | CHEN Kunfeng, HU Qianyu, LIU Feng, XUE Dongfeng. Multi-scale Crystallization Materials: Advances in in-situ Characterization Techniques and Computational Simulations [J]. Journal of Inorganic Materials, 2023, 38(3): 256-269. |
| [13] | LIN Siqi, LI Airan, FU Chenguang, LI Rongbing, JIN Min. Crystal Growth and Thermoelectric Properties of Zintl Phase Mg3X2 (X=Sb, Bi) Based Materials: a Review [J]. Journal of Inorganic Materials, 2023, 38(3): 270-279. |
| [14] | LIU Yan, ZHANG Keying, LI Tianyu, ZHOU Bo, LIU Xuejian, HUANG Zhengren. Electric-field Assisted Joining Technology for the Ceramics Materials: Current Status and Development Trend [J]. Journal of Inorganic Materials, 2023, 38(2): 113-124. |
| [15] | XIE Bing, CAI Jinxia, WANG Tongtong, LIU Zhiyong, JIANG Shenglin, ZHANG Haibo. Research Progress of Polymer-based Multilayer Composite Dielectrics with High Energy Storage Density [J]. Journal of Inorganic Materials, 2023, 38(2): 137-147. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||