无机材料学报 ›› 2023, Vol. 38 ›› Issue (9): 1110-1116.DOI: 10.15541/jim20220756
所属专题: 【材料计算】计算材料(202309); 【能源环境】钙钛矿(202310); 【能源环境】太阳能电池(202310)
• 研究快报 • 上一篇
吴晓维1( ), 张涵1,2, 曾彪1,2, 明辰1,2, 孙宜阳1,2()
), 张涵1,2, 曾彪1,2, 明辰1,2, 孙宜阳1,2()
收稿日期:2022-12-17
修回日期:2023-05-11
出版日期:2023-09-20
网络出版日期:2023-06-01
通讯作者:
孙宜阳, 研究员. E-mail: yysun@mail.sic.ac.cn作者简介:吴晓维(1993-), 女, 硕士. E-mail: wxw_xiaowei@163.com
WU Xiaowei1(), ZHANG Han1,2, ZENG Biao1,2, MING Chen1,2, SUN Yiyang1,2()
Received:2022-12-17
Revised:2023-05-11
Published:2023-09-20
Online:2023-06-01
Contact:
SUN Yiyang, professor. E-mail: yysun@mail.sic.ac.cnAbout author:WU Xiaowei (1993-), female, Master. E-mail: wxw_xiaowei@163.com
Supported by:摘要:
在卤族钙钛矿材料的缺陷研究中, 密度泛函理论计算发挥着重要作用。传统的半局域泛函(如PBE)虽然能够得到与实验接近的禁带宽度, 但是已有研究表明其不能准确描述材料的带边位置。采用更准确的杂化泛函, 结合自旋轨道耦合(SOC)效应与充分的结构优化开展缺陷研究十分必要。可以选择两种杂化泛函, 即屏蔽的杂化泛函HSE和非屏蔽的杂化泛函PBE0。本研究以正交相CsPbI3为例, 系统比较了两种方法在缺陷性质计算上的差异。计算结果表明, 对于体相性质, 两种杂化泛函并无明显的差别。但是, 对于缺陷性质, 两种泛函出现定性的差别。HSE计算中预测的浅能级缺陷, 在PBE0计算中大部分变为深能级缺陷, 且缺陷转变能级和Kohn-Sham能级均出现定性差别。上述差别的本质在于, Hartree-Fock交换势具有长程作用特征, 因而普通的杂化泛函如PBE0在计算量允许的超胞尺寸上无法得到收敛的结果, 而HSE对上述交换势具有屏蔽作用, 可采用相对小尺寸的超胞得到收敛的缺陷能级。本研究结果表明, 尽管HSE杂化泛函需要较大的Hartree-Fock混合参数(约0.43), 其仍是准确计算卤族钙钛矿缺陷性质的有效方法。
中图分类号:
吴晓维, 张涵, 曾彪, 明辰, 孙宜阳. 杂化泛函HSE和PBE0计算CsPbI3缺陷性质的比较研究[J]. 无机材料学报, 2023, 38(9): 1110-1116.
WU Xiaowei, ZHANG Han, ZENG Biao, MING Chen, SUN Yiyang. Comparison of Hybrid Functionals HSE and PBE0 in Calculating the Defect Properties of CsPbI3[J]. Journal of Inorganic Materials, 2023, 38(9): 1110-1116.
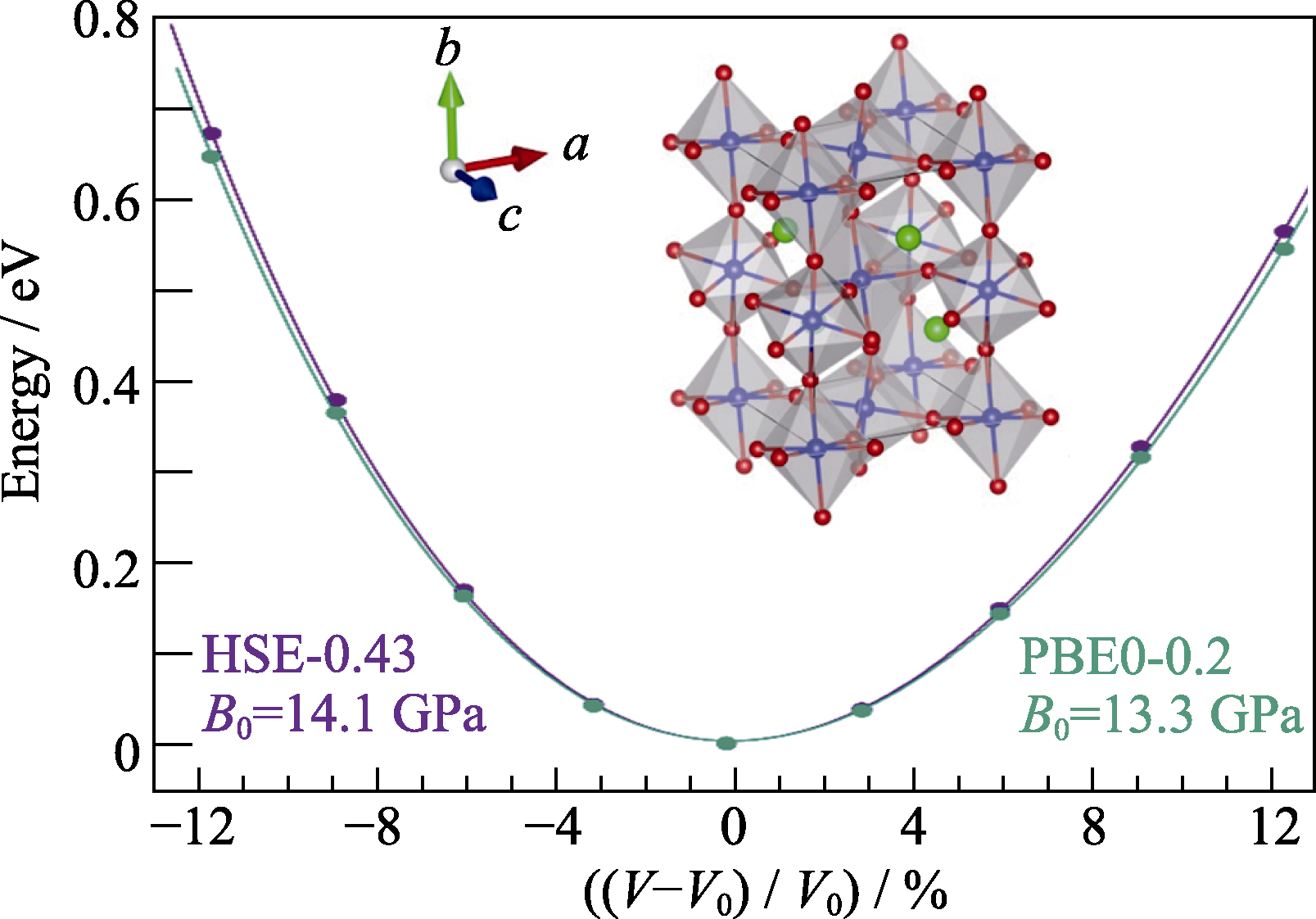
Fig. 1 Fitting Murnaghan equation of state to obtain the equilibrium volume and bulk modulus with inset showing the atomic structure of γ-phase CsPbI3
| HSE-0.43 | PBE0-0.20 | |||||
|---|---|---|---|---|---|---|
| X/a | Y/b | Z/c | X/a | Y/b | Z/c | |
| a/nm 0.9008 | b/nm 1.2525 | c/nm 0.8632 | a/nm 0.9061 | b/nm 1.2589 | c/nm 0.8674 | |
| Cs | 0.4312 | 0.25 | 0.0228 | 0.4294 | 0.25 | 0.0238 |
| Pb | 0 | 0 | 0 | 0 | 0 | 0 |
| I1 | 0.5110 | 0.25 | 0.5783 | 0.5104 | 0.25 | 0.5804 |
| I2 | 0.2021 | 0.0387 | 0.3017 | 0.2017 | 0.0395 | 0.3021 |
Table 1 Lattice constants and internal parameters of orthorhombic CsPbI3 calculated by two different hybrid functionals
| HSE-0.43 | PBE0-0.20 | |||||
|---|---|---|---|---|---|---|
| X/a | Y/b | Z/c | X/a | Y/b | Z/c | |
| a/nm 0.9008 | b/nm 1.2525 | c/nm 0.8632 | a/nm 0.9061 | b/nm 1.2589 | c/nm 0.8674 | |
| Cs | 0.4312 | 0.25 | 0.0228 | 0.4294 | 0.25 | 0.0238 |
| Pb | 0 | 0 | 0 | 0 | 0 | 0 |
| I1 | 0.5110 | 0.25 | 0.5783 | 0.5104 | 0.25 | 0.5804 |
| I2 | 0.2021 | 0.0387 | 0.3017 | 0.2017 | 0.0395 | 0.3021 |
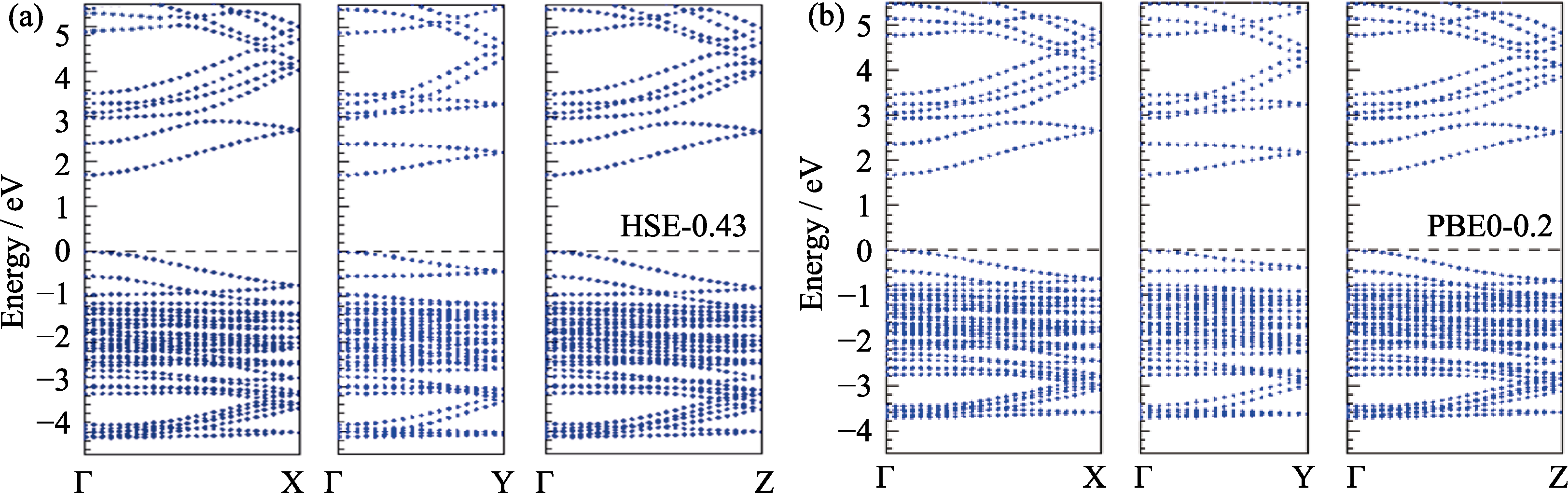
Fig. 2 Band structures of γ-CsPbI3 calculated by two hybrid functionals HSE-0.43 (a) and PBE0-0.2 (b) including the SOC effect
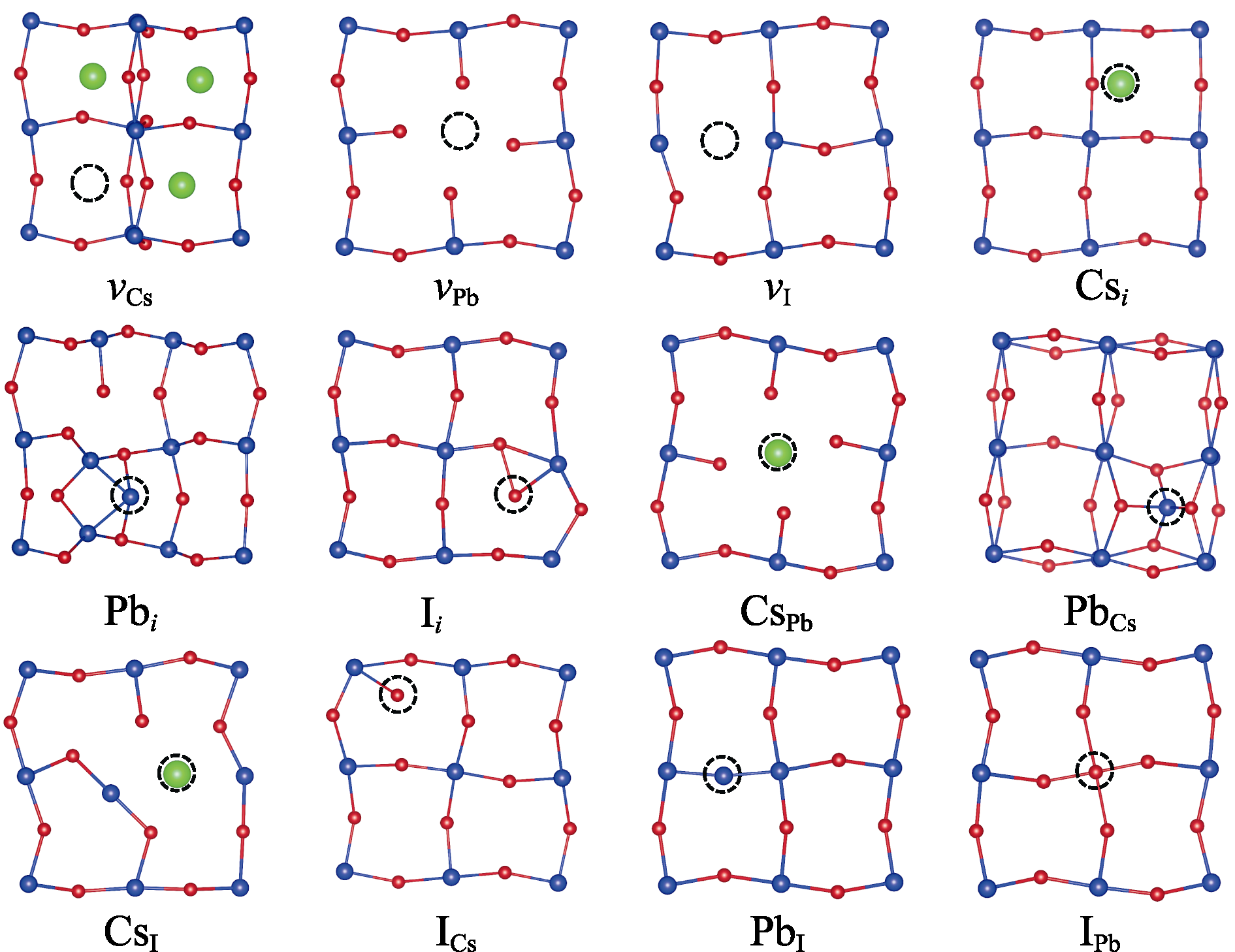
Fig. 3 Atomic structures of 12 intrinsic defects after relaxation in neutral charge state As two functionals yield similar structures, only the structures from PBE0-0.2 calculations are shown here
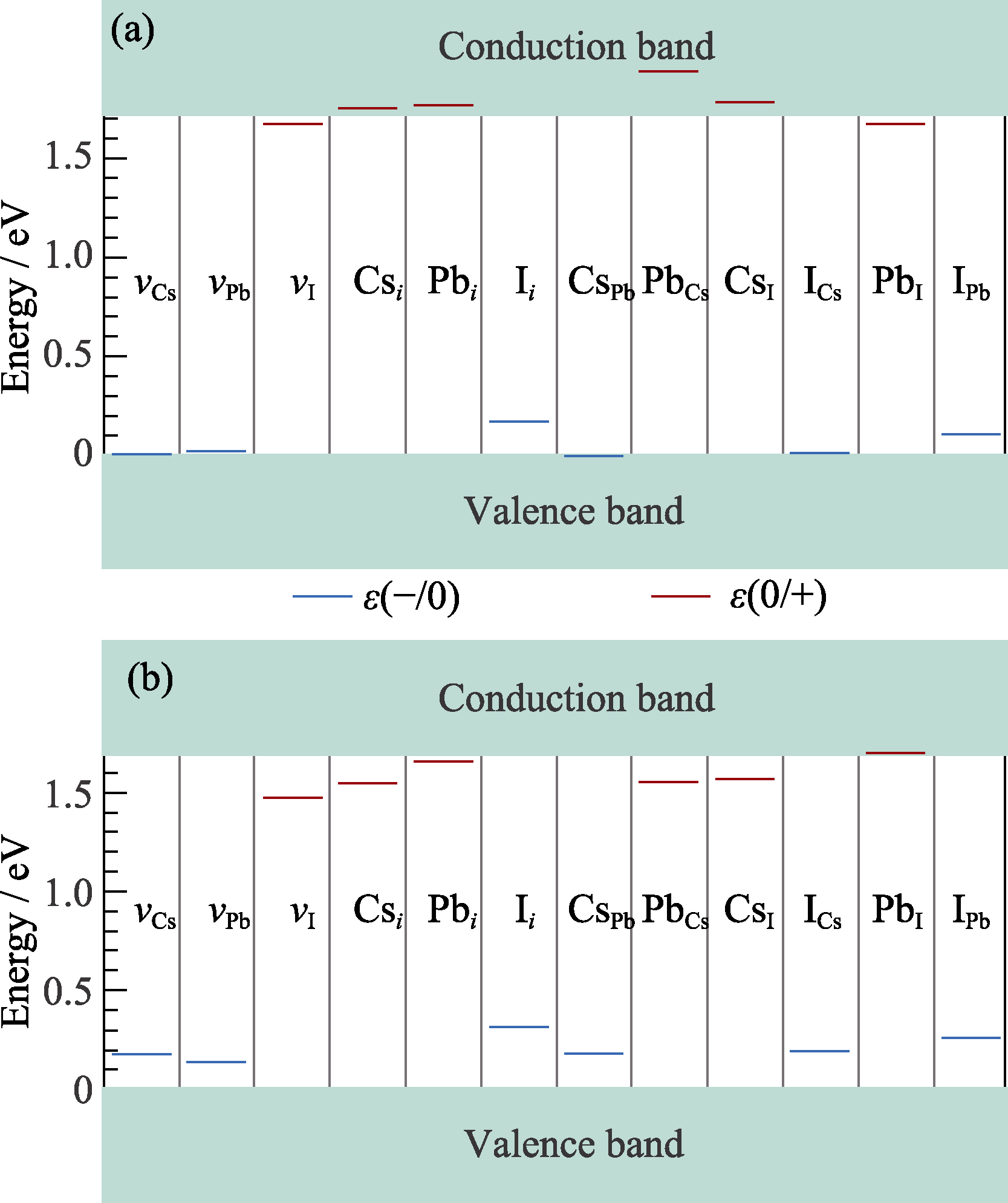
Fig. 4 Defect transition levels in γ-CsPbI3 calculated by HSE-0.43 (a) and PBE0-0.2 (b) Blue lines: acceptor levels; Red lines: donor levels
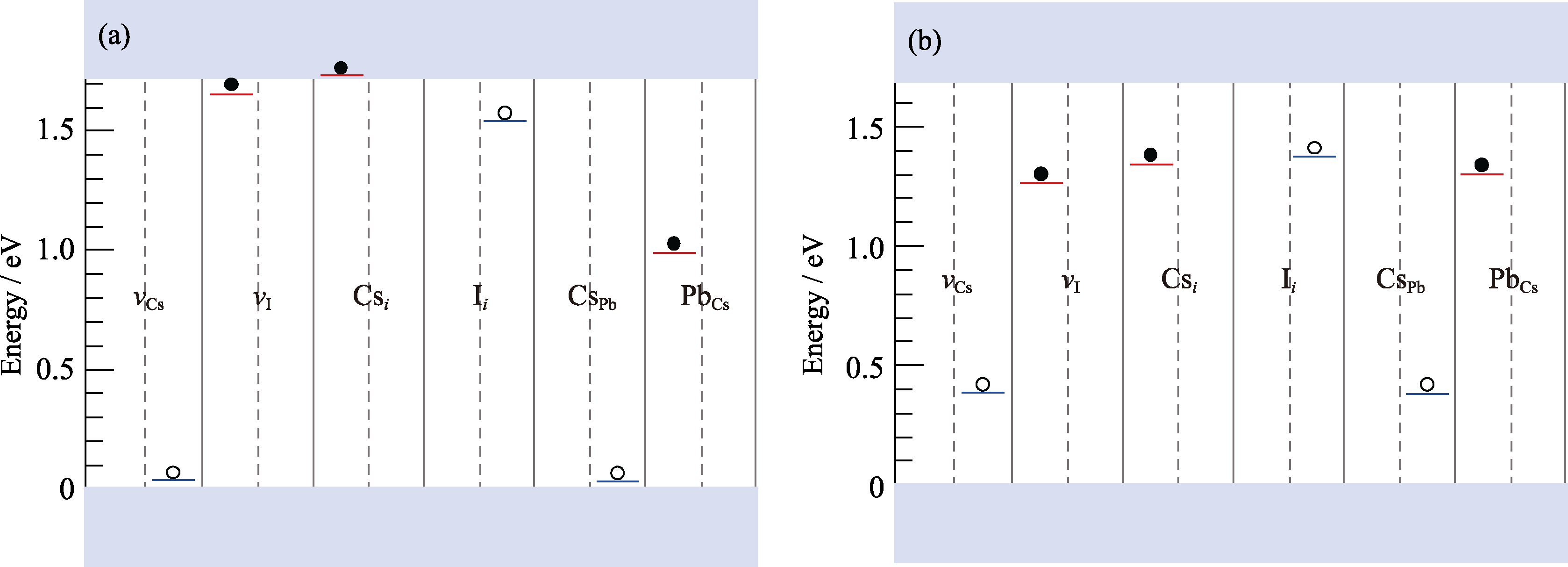
Fig. 5 Kohn-Sham energy levels of vCs, vI, Csi, Ii, CsPb and PbCs defects calculated by HSE-0.43 (a) and PBE0-0.2 (b) For acceptor defects, the left half is for −1 state, while the right half is for the neutral state. For donor defects, the left half is for neutral state, while the right half is for +1 state. Open and solid circles represent holes and electrons, respectively.
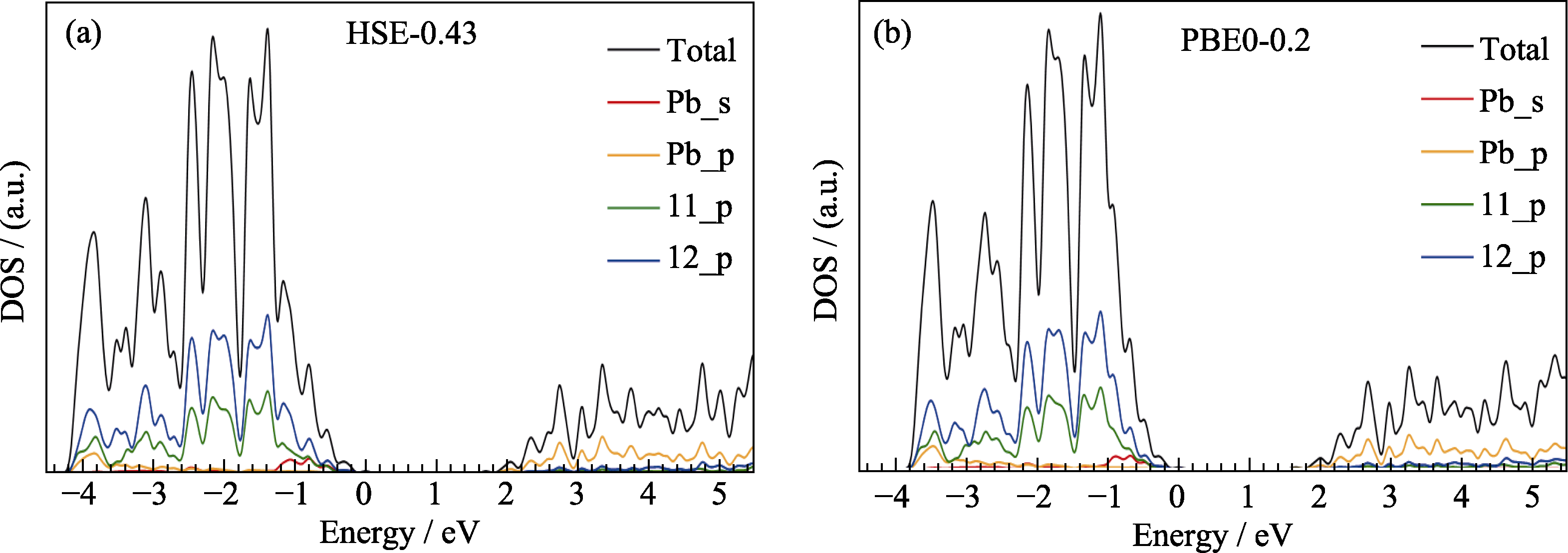
Fig. S1 Total and projected density of states of γ-CsPbI3 calculated by two hybrid functionals HSE-0.43 (a) and PBE0- 0.2 (b) including the SOC effect
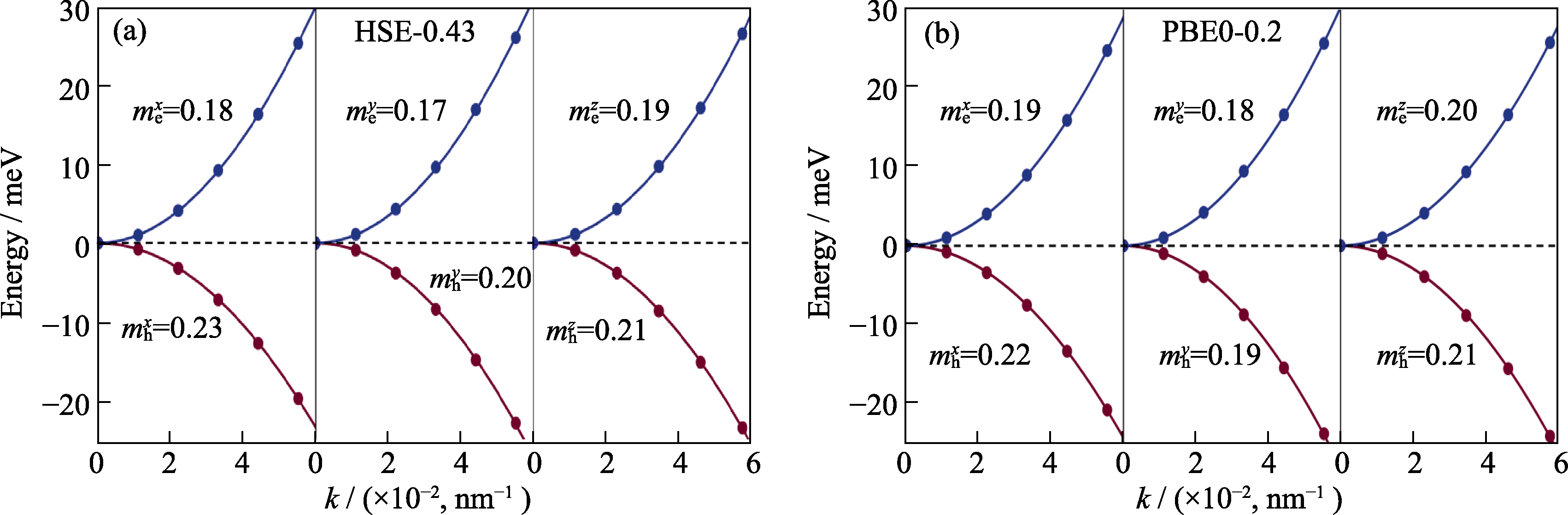
Fig. S2 Parabolic fitting of the band near Γ point to obtained the effective masses of electrons and holes. For clarity, the electron and hole bands are referenced to the CBM and VBM, respectively; SOC effect is included here; (a) HSE-0.43; (b) PBE0-0.2
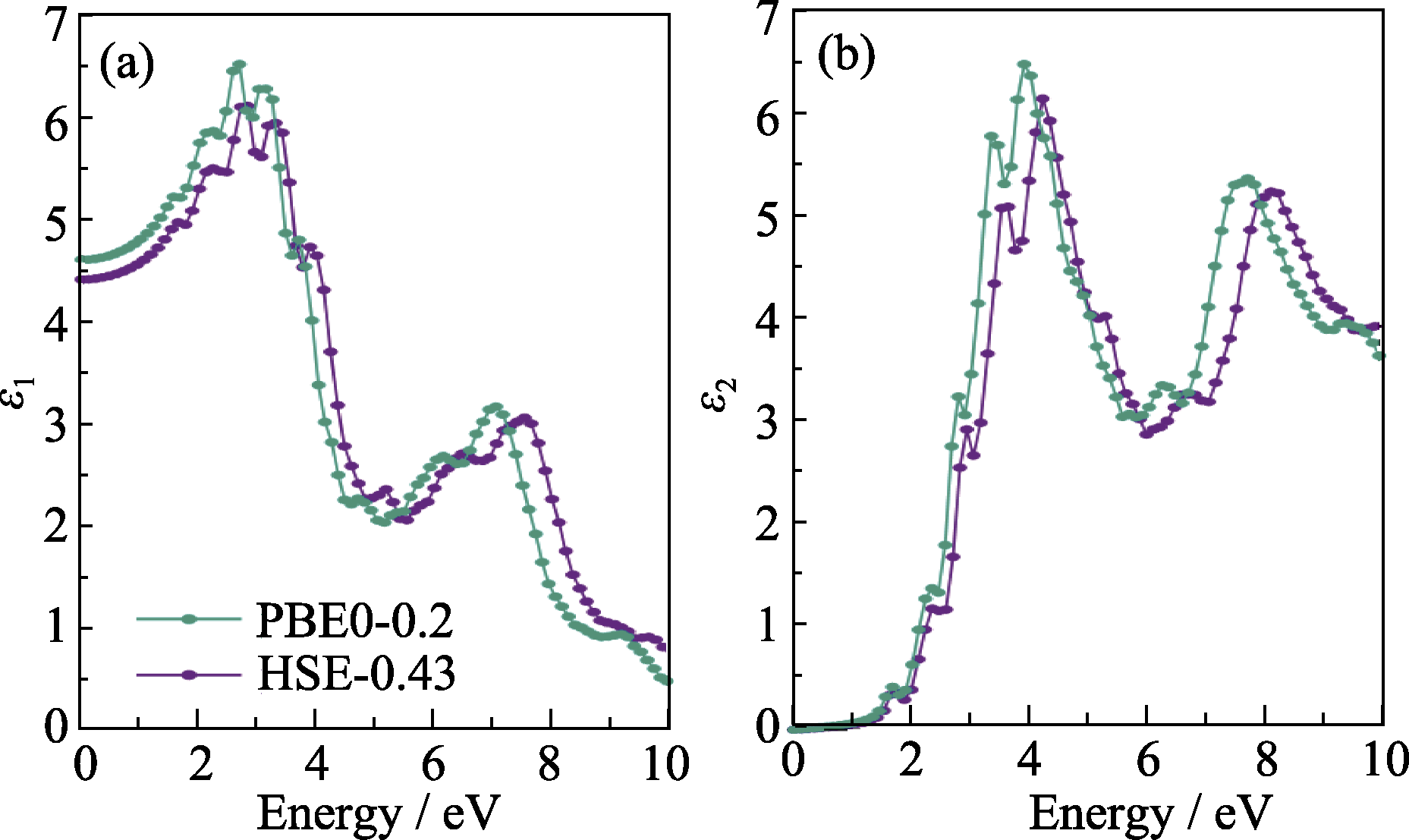
Fig. S3 Comparison of dielectric constants calculated by two hybrid functionals (a) Real part ε1; (b) Imaginary part ε2; Calculations considered SOC effect and employed 5×4×5 k-grid and 544 empty bands
| [1] |
LEE M M, TEUSCHER J, MIYASAKA T, et al. Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science, 2012, 338: 643.
DOI PMID |
| [2] |
BURSCHKA J, PELLET N, MOON S J, et al. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature, 2013, 499(7458): 316.
DOI |
| [3] |
GREEN M A, HO-BAILLIE A, SNAITH H J. The emergence of perovskite solar cells. Nature Photonics, 2014, 8(7): 506.
DOI |
| [4] |
JEON N J, NOH J H, YANG W S, et al. Compositional engineering of perovskite materials for high-performance solar cells. Nature, 2015, 517(7535): 476.
DOI |
| [5] |
SALIBA M, MATSUI T, DOMANSKI K, et al. Incorporation of rubidium cations into perovskite solar cells improves photovoltaic performance. Science, 2016, 354(6309): 206.
PMID |
| [6] |
MCMEEKIN D P, SADOUGHI G, REHMAN W, et al. A mixed- cation lead mixed-halide perovskite absorber for tandem solar cells. Science, 2016, 351(6269): 151.
DOI URL |
| [7] |
KOJIMA A, TESHIMA K, SHIRAI Y, et al. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. Journal of the American Chemical Society, 2009, 131: 6050.
DOI PMID |
| [8] |
LIU M, JOHNSTON M B, SNAITH H J. Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature, 2013, 501(7467): 395.
DOI |
| [9] | NREL. Best research-cell efficiency chart[2023-04-05]. https://www.nrel.gov/pv/cell-efficiency.html. |
| [10] | BELIKOVICH B A, PASHCHUK I P, PIDZYRAILO N S. Luminescence of CsPbCl3 single-crystals. Optikai I Spectroskopiya, 1977, 42: 113. |
| [11] |
ERA M, MORIMOTO S, TSUTSUI T, et al. Organic-inorganic heterostructure electroluminescent device using a layered perovskite semiconductor (C6H5C2H4NH3)2PbI4. Applied Physics Letters, 1994, 65(6): 676.
DOI URL |
| [12] |
TAN Z K, MOGHADDAM R S, LAI M L, et al. Bright light-emitting diodes based on organometal halide perovskite. Nature Nanotechnology, 2014, 9(9): 687.
DOI |
| [13] |
LIN K, XING J, QUAN L N, et al. Perovskite light-emitting diodes with external quantum efficiency exceeding 20 percent. Nature, 2018, 562(7726): 245.
DOI |
| [14] |
CAO Y, WANG N, TIAN H, et al. Perovskite light-emitting diodes based on spontaneously formed submicrometre-scale structures. Nature, 2018, 562(7726): 249.
DOI |
| [15] |
STRANKS S D, SNAITH H J. Metal-halide perovskites for photovoltaic and light-emitting devices. Nature Nanotechnology, 2015, 10(5): 391.
DOI PMID |
| [16] |
FANG Y, DONG Q, SHAO Y, et al. Highly narrowband perovskite single-crystal photodetectors enabled by surface-charge recombination. Nature Photonics, 2015, 9(10): 679.
DOI |
| [17] |
DOU L, YANG Y M, YOU J, et al. Solution-processed hybrid perovskite photodetectors with high detectivity. Nature Communications, 2014, 5: 5404.
DOI PMID |
| [18] |
YIN W J, SHI T, YAN Y. Unusual defect physics in CH3NH3Pb3 perovskite solar cell absorber. Applied Physics Letters, 2014, 104(6): 063903.
DOI URL |
| [19] |
YIN W, SHI T, YAN Y. Unique properties of halide perovskites as possible origins of the superior solar cell performance. Advanced Materials, 2014, 26(27): 4653.
DOI URL |
| [20] |
BRANDT R E, STEVANOVIĆ V, GINLEY D S, et al. Identifying defect-tolerant semiconductors with high minority-carrier lifetimes: beyond hybrid lead halide perovskites. MRS Communications, 2015, 5(2): 265.
DOI URL |
| [21] |
MEGGIOLARO D, MOTTI S G, MOSCONI E, et al. Iodine chemistry determines the defect tolerance of lead-halide perovskites. Energy and Environmental Science, 2018, 11(3): 702.
DOI URL |
| [22] |
KURCHIN R C, GORAI P, BUONASSISI T, et al. Structural and chemical features giving rise to defect tolerance of binary semiconductors. Chemistry of Materials, 2018, 30(16): 5583.
DOI URL |
| [23] |
PARK J S, KIM S, XIE Z, et al. Point defect engineering in thin-film solar cells. Nature Reviews Materials, 2018, 3(7): 194.
DOI |
| [24] |
KANG J, WANG L. High defect tolerance in lead halide perovskite CsPbBr3. The Journal of Physical Chemistry Letters, 2017, 8: 489.
DOI URL |
| [25] |
MING C, WANG H, WEST D, et al. Defect tolerance in CsPbI3: reconstruction of potential energy landscape and band degeneracy in spin-orbit coupling. Journal of Materials Chemistry A, 2022, 10(6): 3018.
DOI URL |
| [26] |
AGIORGOUSIS M L, SUN Y Y, ZENG H, et al. Strong covalency-induced recombination centers in perovskite solar cell material CH3NH3PbI3. Journal of the American Chemical Society, 2014, 136(41): 14570.
DOI PMID |
| [27] |
WU X W, MING C, SHI J, et al. Defects in statically unstable solids: the case for cubic perovskite α-CsPbI3. Chinese Physics Letters, 2022, 39(4): 046101.
DOI |
| [28] |
NEUKIRCH A J, NIE W, BLANCON J C, et al. Polaron stabilization by cooperative lattice distortion and cation rotations in hybrid perovskite materials. Nano Letters, 2016, 16(6): 3809.
DOI PMID |
| [29] |
GAO Y, LUO T, XIA Y, et al. The joint effect of spin-orbit coupling and atomistic disorder on bandgap evolution in inorganic CsSn1-xPbxI3 mixed perovskite. Journal of Applied Physics, 2022, 131(5): 055107.
DOI URL |
| [30] |
MOSCONI E, MERABET B, MEGGIOLARO D, et al. First-principles modeling of bismuth doping in the MAPbI3 perovskite. Journal of Physical Chemistry C, 2018, 122(25): 14107.
DOI URL |
| [31] |
BISCHOFF T, WIKTOR J, CHEN W, et al. Nonempirical hybrid functionals for band gaps of inorganic metal-halide perovskites. Physical Review Materials, 2019, 3(12): 123802.
DOI URL |
| [32] |
TAO S, SCHMIDT I, BROCKS G, et al. Absolute energy level positions in tin- and lead-based halide perovskites. Nature Communications, 2019, 10: 2560.
DOI PMID |
| [33] |
MAHATA A, MEGGIOLARO D, DE ANGELIS F. From large to small polarons in lead, tin, and mixed lead-tin halide perovskites. Journal of Physical Chemistry Letters, 2019, 10(8): 1790.
DOI PMID |
| [34] |
WANG J, ZHANG J, ZHOU Y, et al. Highly efficient all-inorganic perovskite solar cells with suppressed non-radiative recombination by a lewis base. Nature Communications, 2020, 11: 177.
DOI PMID |
| [35] |
LIU C, IGCI C, YANG Y, et al. Dopant-free hole transport materials afford efficient and stable inorganic perovskite solar cells and modules. Angewandte Chemie International Edition, 2021, 60(37): 20489.
DOI URL |
| [36] |
KAISER W, CARIGNANO M, ALOTHMAN A A, et al. First-principles molecular dynamics in metal-halide perovskites: contrasting generalized gradient approximation and hybrid functionals. Journal of Physical Chemistry Letters, 2021, 12(49): 11886.
DOI PMID |
| [37] |
VONA C, NABOK D, DRAXL C. Electronic structure of (organic-)inorganic metal halide perovskites: the dilemma of choosing the right functional. Advanced Theory and Simulations, 2022, 5(1): 2100496.
DOI URL |
| [38] |
MEGGIOLARO D, DE ANGELIS F. First-principles modeling of defects in lead halide perovskites: best practices and open issues. ACS Energy Letters, 2018, 3(9): 2206.
DOI URL |
| [39] |
EPERON G E, PATERNÒ G M, SUTTON R J, et al. Inorganic caesium lead iodide perovskite solar cells. Journal of Materials Chemistry A, 2015, 3(39): 19688.
DOI URL |
| [40] |
SWARNKAR A, MARSHALL A R, SANEHIRA E M, et al. Quantum dot-induced phase stabilization of α-CsPbI3 perovskite for high-efficiency photovoltaics. Science, 2016, 354(6308): 92.
DOI URL |
| [41] |
YOON S M, MIN H, KIM J B, et al. Surface engineering of ambient-air-processed cesium lead triiodide layers for efficient solar cells. Joule, 2021, 5: 183.
DOI URL |
| [42] |
WANG P, ZHANG X, ZHOU Y, et al. Solvent-controlled growth of inorganic perovskite films in dry environment for efficient and stable solar cells. Nature Communications, 2018, 9: 2225.
DOI |
| [43] |
WANG Y, DAR M I, ONO L K, et al. Thermodynamically stabilized β-CsPbI3-based perovskite solar cells with efficiencies > 18%. Science, 2019, 365(6453): 591.
DOI URL |
| [44] |
ZHAO B, JIN S F, HUANG S, et al. Thermodynamically stable orthorhombic γ-CsPbI3 thin films for high-performance photovoltaics. Journal of the American Chemical Society, 2018, 140(37): 11716.
DOI URL |
| [45] |
WANG K, JIN Z, LIANG L, et al. Chlorine doping for black γ-CsPbI3 solar cells with stabilized efficiency beyond 16%. Nano Energy, 2019, 58: 175.
DOI URL |
| [46] |
LI Z, YANG M, PARK J S, et al. Stabilizing perovskite structures by tuning tolerance factor: formation of formamidinium and cesium lead iodide solid-state alloys. Chemistry of Materials, 2016, 28(1): 284.
DOI URL |
| [47] |
WANG Y, CHEN Y, ZHANG T, et al. Chemically stable black phase CsPbI3 inorganic perovskites for high-efficiency photovoltaics. Advanced Materials, 2020, 32(45): 2001025.
DOI URL |
| [48] | XIANG W, LIU S, TRESS W, et al. A review on the stability of inorganic metal halide perovskites: challenges and opportunities for stable solar cells. Energy & Environmental Science, 2021, 14(4): 2090. |
| [49] |
CUI Y, SHI J, MENG F, et al. A versatile molten-salt induction strategy to achieve efficient CsPbI3 perovskite solar cells with a high open-circuit voltage >1.2 V. Advanced Materials, 2022, 34(45): 2205028.
DOI URL |
| [50] |
KRESSE G, FURTHMÜLLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane- wave basis set. Computational Materials Science, 1996, 6: 15.
DOI URL |
| [51] |
BLÖCHL P. E. Projector augmented-wave method. Physical Review B, 1994, 50(24): 17953.
DOI PMID |
| [52] |
KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method. Physical Review B, 1999, 59: 1758.
DOI URL |
| [53] |
WU X W, GAO W, CHAI J, et al. Defect tolerance in chalcogenide perovskite photovoltaic material BaZrS3. Science China Materials, 2021, 64(12): 2976.
DOI |
| [54] |
ZHANG X, TURIANSKY M E, SHEN J X, et al. Iodine interstitials as a cause of nonradiative recombination in hybrid perovskites. Physical Review B, 2020, 101(14): 140101(R).
DOI URL |
| [55] |
BANG J, SUN Y Y, ABTEW T A, et al. Difficulty in predicting shallow defects with hybrid functionals: implication of the long-range exchange interaction. Physical Review B, 2013, 88(3): 035134.
DOI URL |
| [1] | 董思吟, 帖舒婕, 袁瑞涵, 郑霄家. 低维卤化物钙钛矿直接型X射线探测器研究进展[J]. 无机材料学报, 2023, 38(9): 1017-1030. |
| [2] | 王润, 相恒阳, 曾海波. 钙钛矿多色级联发光二极管中多中心载流子均衡分布调控研究[J]. 无机材料学报, 2023, 38(9): 1062-1068. |
| [3] | 王马超, 唐扬敏, 邓明雪, 周真真, 刘小峰, 王家成, 刘茜. 共沉淀法制备Cs2Ag0.1Na0.9BiCl6:Tm3+双钙钛矿及其近红外发光性能[J]. 无机材料学报, 2023, 38(9): 1083-1088. |
| [4] | 韩旭, 姚恒大, 吕梅, 陆红波, 朱俊. 单分子液晶添加剂在甲脒铅碘钙钛矿太阳能电池中的应用[J]. 无机材料学报, 2023, 38(9): 1097-1102. |
| [5] | 方万丽, 沈黎丽, 李海艳, 陈薪羽, 陈宗琦, 寿春晖, 赵斌, 杨松旺. NiOx介孔层的成膜过程对碳电极钙钛矿太阳能电池性能的影响[J]. 无机材料学报, 2023, 38(9): 1103-1109. |
| [6] | 蔡凯, 靳志文. 基于二维钙钛矿(PEA)2PbI4的光电探测器[J]. 无机材料学报, 2023, 38(9): 1069-1075. |
| [7] | 丁统顺, 丰平, 孙学文, 单沪生, 李琪, 宋健. Fmoc-FF-OH钝化钙钛矿薄膜及其太阳能电池性能研究[J]. 无机材料学报, 2023, 38(9): 1076-1082. |
| [8] | 张伦, 吕梅, 朱俊. Cs2AgBiBr6钙钛矿太阳能电池研究进展[J]. 无机材料学报, 2023, 38(9): 1044-1054. |
| [9] | 陈雨, 林埔安, 蔡冰, 张文华. 钙钛矿太阳能电池无机空穴传输材料的研究进展[J]. 无机材料学报, 2023, 38(9): 991-1004. |
| [10] | 郭华军, 安帅领, 孟婕, 任书霞, 王文文, 梁子尚, 宋佳钰, 陈恒彬, 苏航, 赵晋津. 卤化物钙钛矿光电阻变机理研究进展[J]. 无机材料学报, 2023, 38(9): 1005-1016. |
| [11] | 代晓栋, 张露伟, 钱奕成, 任智鑫, 曹焕奇, 印寿根. 锡铅混合钙钛矿太阳能电池垂直组分梯度的溶剂工程调控[J]. 无机材料学报, 2023, 38(9): 1089-1096. |
| [12] | 李俊生, 曾良, 刘荣军, 王衍飞, 万帆, 李端. 锶钽氧氮化物功能陶瓷的高效合成、致密化及介电性能研究[J]. 无机材料学报, 2023, 38(8): 885-892. |
| [13] | 樊家顺, 夏冬林, 刘保顺. 温度相关的CsPbBr3纳米晶瞬态光电导响应研究[J]. 无机材料学报, 2023, 38(8): 893-900. |
| [14] | 杨颖康, 邵怡晴, 李柏良, 吕志伟, 王路路, 王亮君, 曹逊, 吴宇宁, 黄荣, 杨长. Cl掺杂对CuI薄膜发光性能增强研究[J]. 无机材料学报, 2023, 38(6): 687-692. |
| [15] | 张万文, 罗建强, 刘淑娟, 马建国, 张小平, 杨松旺. 氧化锆间隔层的低温喷涂制备及其三层结构钙钛矿太阳能电池应用性能[J]. 无机材料学报, 2023, 38(2): 213-218. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||