无机材料学报 ›› 2021, Vol. 36 ›› Issue (11): 1125-1136.DOI: 10.15541/jim20200683
所属专题: 【虚拟专辑】电致变色与热致变色材料
• 综述 • 下一篇
赵林艳1( ), 刘阳思1,2,3, 席晓丽1,2,4(), 马立文1,2, 聂祚仁1,2,4
), 刘阳思1,2,3, 席晓丽1,2,4(), 马立文1,2, 聂祚仁1,2,4
收稿日期:2020-11-28
修回日期:2021-04-26
出版日期:2021-11-20
网络出版日期:2021-06-01
通讯作者:
席晓丽, 教授. E-mail: xixiaoli@bjut.edu.cn
作者简介:赵林艳(1992-), 女, 博士研究生. E-mail: zlyding@emails.bjut.edu.cn
基金资助:
ZHAO Linyan1(), LIU Yangsi1,2,3, XI Xiaoli1,2,4(), MA Liwen1,2, NIE Zuoren1,2,4
Received:2020-11-28
Revised:2021-04-26
Published:2021-11-20
Online:2021-06-01
Contact:
XI Xiaoli, professor. E-mail: xixiaoli@bjut.edu.cn
About author:ZHAO Linyan(1992-), female, PhD candidate. E-mail: zlyding@emails.bjut.edu.cn
Supported by:摘要:
纳米氧化钨作为一种具有独特物理化学性质的半导体功能材料, 已被广泛应用于环境、能源、生命科学、信息技术等领域。本文基于第一性原理计算在纳米氧化钨中的应用进展, 概述了量子力学基础上的第一性原理及密度泛函理论的发展历程及基本理论, 介绍了该领域常用的MS (Materials studio)、VASP (Vienna ab initio simulation package)等模拟计算软件, 并分类阐述了第一性原理计算对氧化钨的微观电子结构、物质相互作用、分子热动力学等方面的研究成果。最后提出了第一性原理计算在纳米氧化钨这类半导体材料研究中存在的问题及未来发展趋势。
中图分类号:
赵林艳, 刘阳思, 席晓丽, 马立文, 聂祚仁. 基于第一性原理计算的纳米氧化钨研究进展[J]. 无机材料学报, 2021, 36(11): 1125-1136.
ZHAO Linyan, LIU Yangsi, XI Xiaoli, MA Liwen, NIE Zuoren. First-principles Study on Nanoscale Tungsten Oxide: a Review[J]. Journal of Inorganic Materials, 2021, 36(11): 1125-1136.
| Type of tungsten oxide | Configuration | 3D Model |
|---|---|---|
| Cubic WO3 | $\text{pm\bar{3}m}\left( 221 \right)$ | |
| Hexagonal WO3 | $\text{p}6/\text{mmm}\left( 191 \right)$ | |
| Tetragonal WO3 | $\text{p}4/\text{ncc}\left( 130 \right)$ | |
| Orthorhombic WO3 | $\text{pbcn}\left( 60 \right)$ | |
| Monoclinic WO3 | \[\text{p}{{2}_{1}}\text{/c}\left( 14 \right)\] | |
| Triclinic WO3 | $\text{p}1\left( 1 \right)$ | |
| Orthorhombic WO2 | $\text{pnma}\left( 62 \right)$ | |
| Monoclinic WO3-x | $\text{p}2/\text{m}\left( 10 \right)$ | |
表1 不同晶型氧化钨的空间结构及3D模型
Table 1 Tungsten oxides with different crystal structures, space groups and 3D models (O and W atoms are represented by red and blue balls, respectively)
| Type of tungsten oxide | Configuration | 3D Model |
|---|---|---|
| Cubic WO3 | $\text{pm\bar{3}m}\left( 221 \right)$ | |
| Hexagonal WO3 | $\text{p}6/\text{mmm}\left( 191 \right)$ | |
| Tetragonal WO3 | $\text{p}4/\text{ncc}\left( 130 \right)$ | |
| Orthorhombic WO3 | $\text{pbcn}\left( 60 \right)$ | |
| Monoclinic WO3 | \[\text{p}{{2}_{1}}\text{/c}\left( 14 \right)\] | |
| Triclinic WO3 | $\text{p}1\left( 1 \right)$ | |
| Orthorhombic WO2 | $\text{pnma}\left( 62 \right)$ | |
| Monoclinic WO3-x | $\text{p}2/\text{m}\left( 10 \right)$ | |
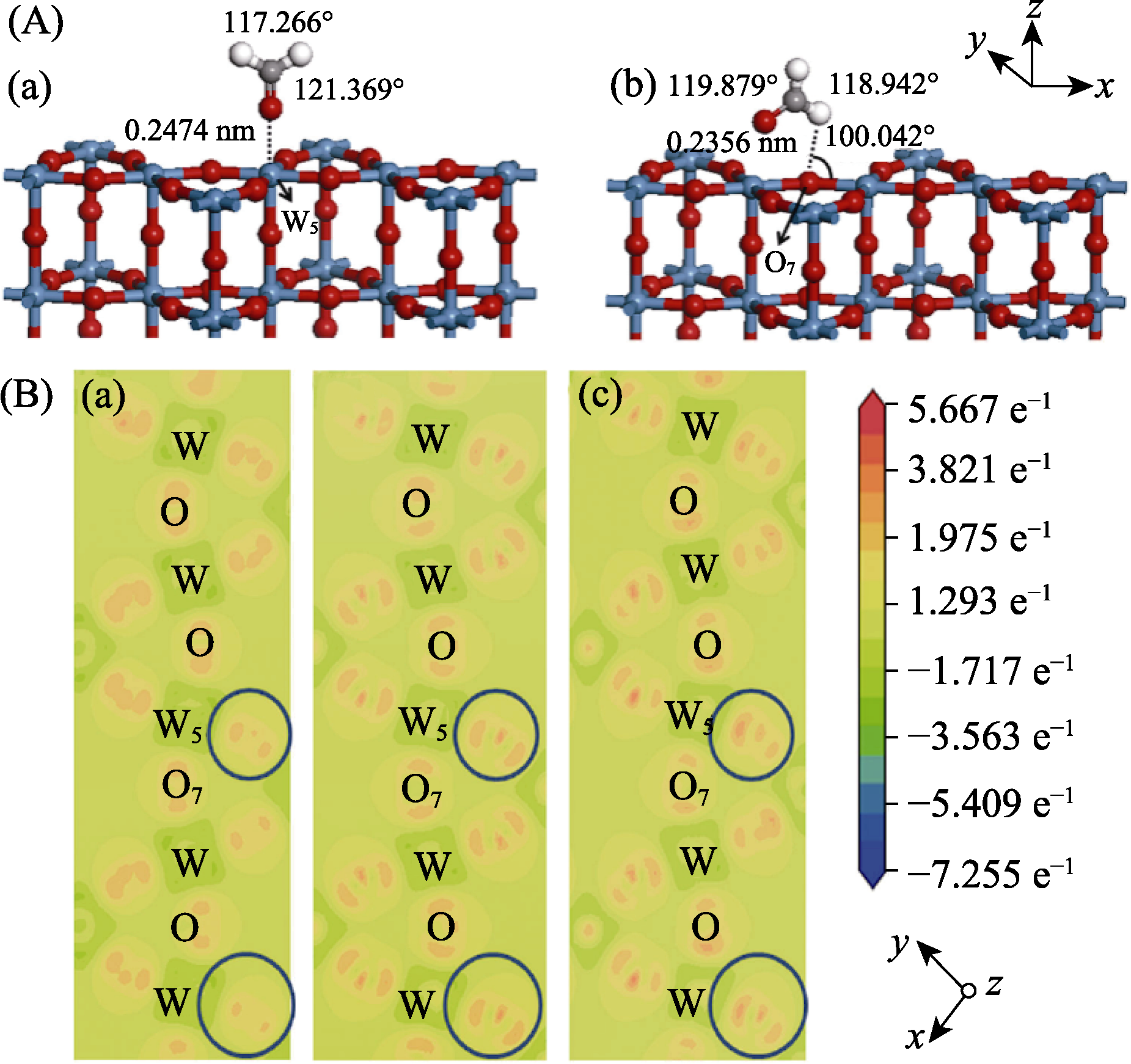
图1 (A)HCHO (红球、白球、黑球分别代表O、H和C原子)在优化后的h-WO3晶体结构(a)W5位点和(b)O7位点吸附; (B)HCHO在h-WO3 (001)面(a)吸附前、(b)吸附于W5位点和(c)吸附于O7位点的模型[59]
Fig. 1 (A) Optimized adsorption structures of HCHO with red, white and black balls representing O, H and C, respectively, on W5 (HCHO-W5 configuration) (a) and O7 (HCOH-O7 configuration) (b) sites of WO-terminated h-WO3 (001) surface; (B) Calculated electron density difference of the clean (001) surface (a), HCHO-absorbed on (001) surface for HCHO-W5 (b) and HCOH-O7 (c) configurations[59]
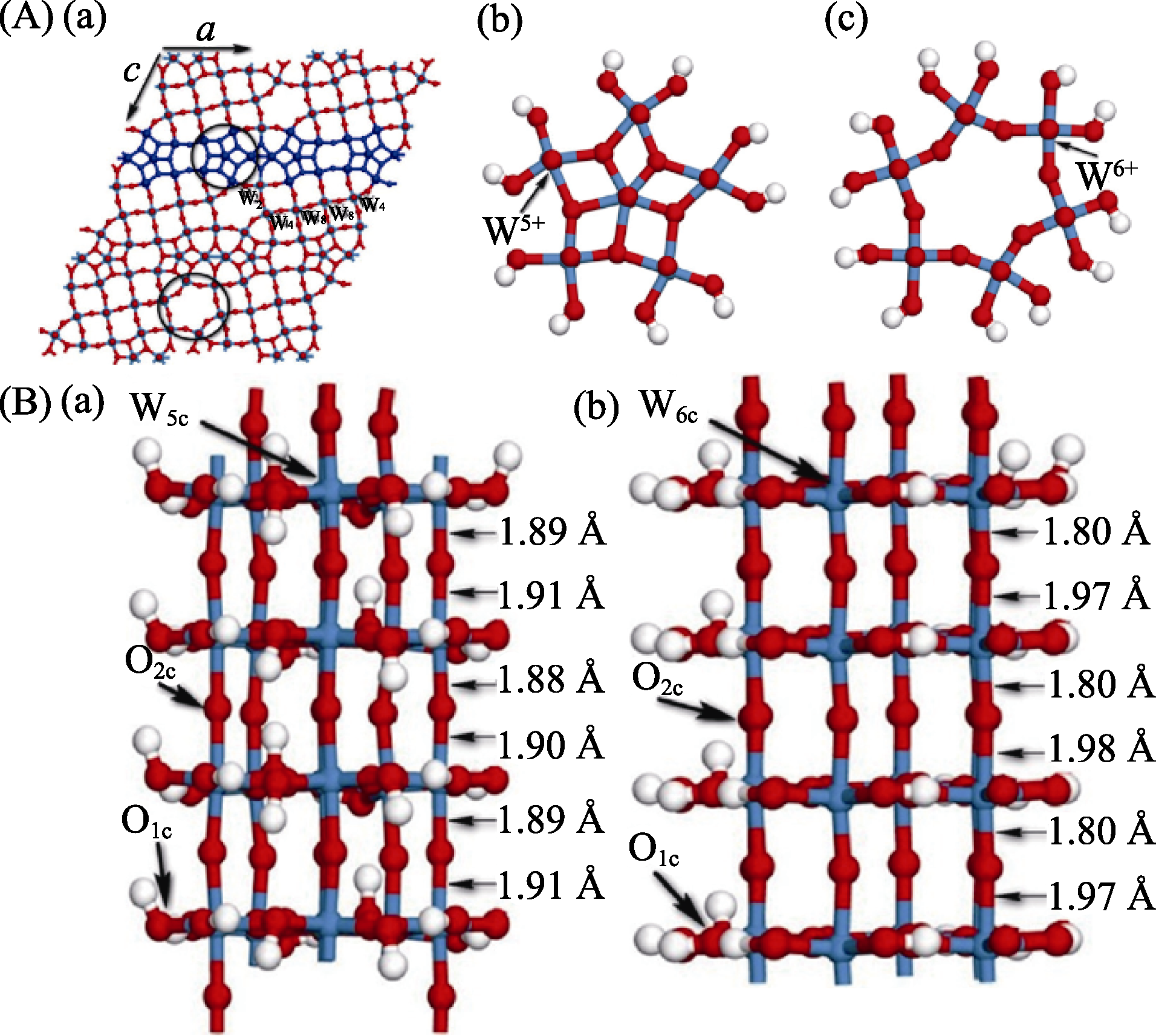
图2 (A)W18O49纳米线超胞模型(a)及从中选出的结构单元 NW1(b)和NW2(c)的俯视图, 其中NW1模型含有的W5+更多, NW2模型含有的W6+更多; (B)W18O49(010)纳米线优化后的NW1(a)和NW2(b)模型及NO2可能的吸附位点[24, 61-62]
Fig. 2 (A) Monoclinic structure (a) of W18O49 nanowires supercell model and its top views of NW1(b) and NW2(c), where NW1 and NW2 include largely cations W5+ and cations W6+, respectively; (B) Optimized models for NW1 (a) and NW2 (b), of W18O49 (010) nanowires[24, 61-62]. O, W and H atoms are represented by red, blue and white balls, respectively (1 Å=0.1 nm)
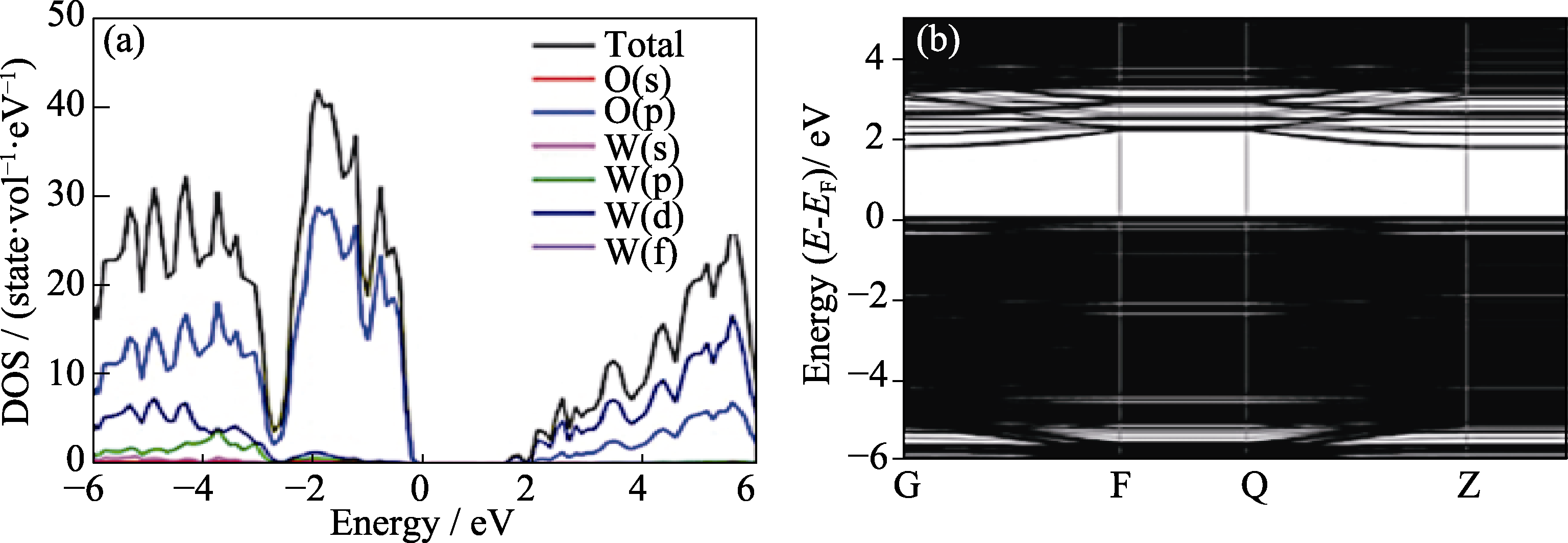
图3 (a)无氧空位的WO3体材料的态密度(DOS)及态密度投影(PDOS)图和(b)具有一个氧空位WO3(002)面的带结构[28]
Fig. 3 (a) Density of states and projected density of states of bulk WO3 without oxygen vacancy, and (b) structure of WO3(002) with one oxygen vacancy[28] Colorful images showing on website
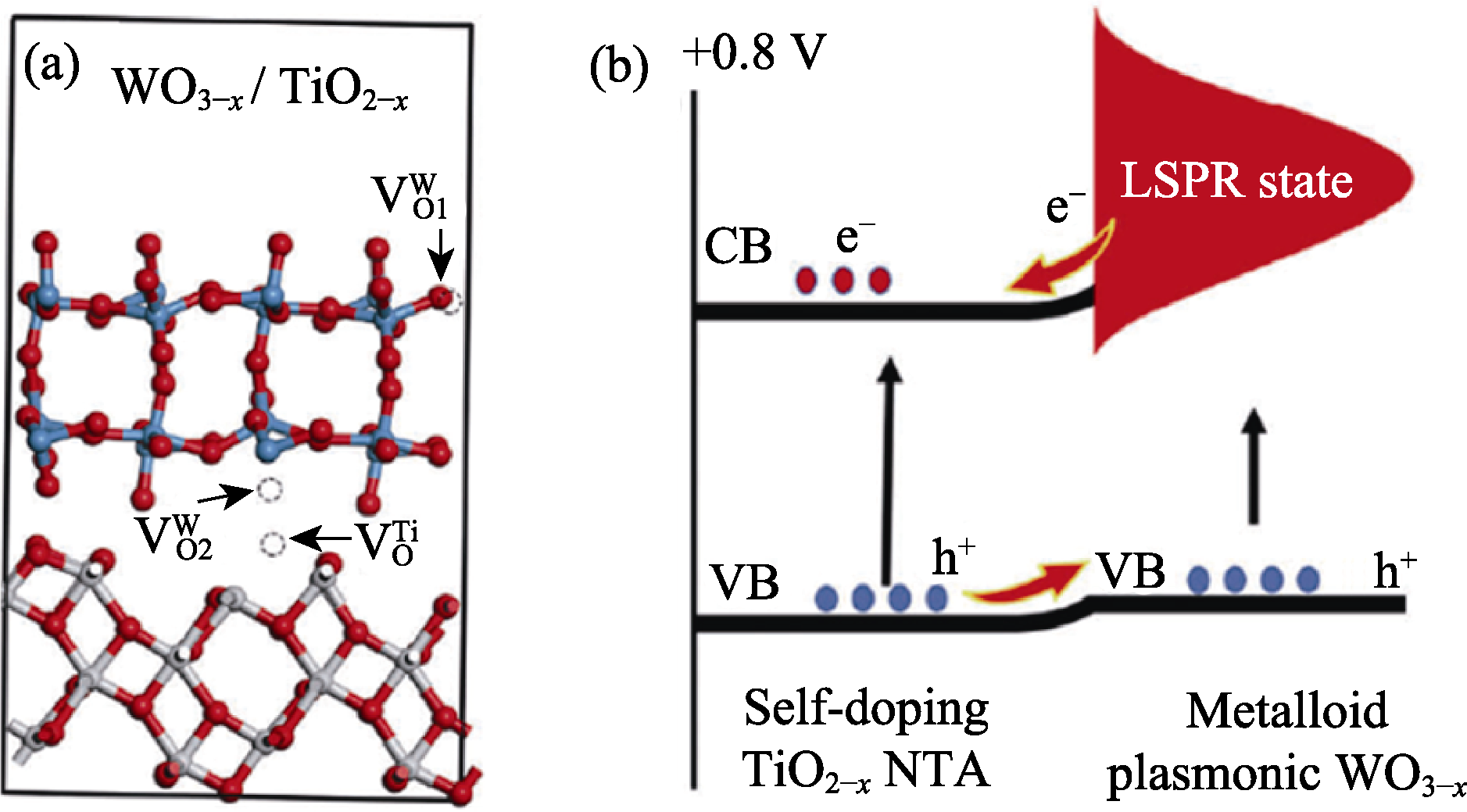
图4 (a)WO3-x/TiO2-x的几何优化平衡结构(红球、蓝球、白球分别代表O、W和Ti原子), 及(b)WO3-x/TiO2-x中的Ti3+自掺杂, 表面等离子激元效应(LSPR)及电荷迁移示意图[72]
Fig. 4 (a) Geometrical optimized equilibrium configuration of WO3-x/TiO2-x with red, blue and white balls representing O, W and Ti, respectively, and (b) schematic diagram of the self-doping Ti3+, localized surface plasmon resonance (LSPR), and charge transfer in WO3-x/TiO2-x[72]
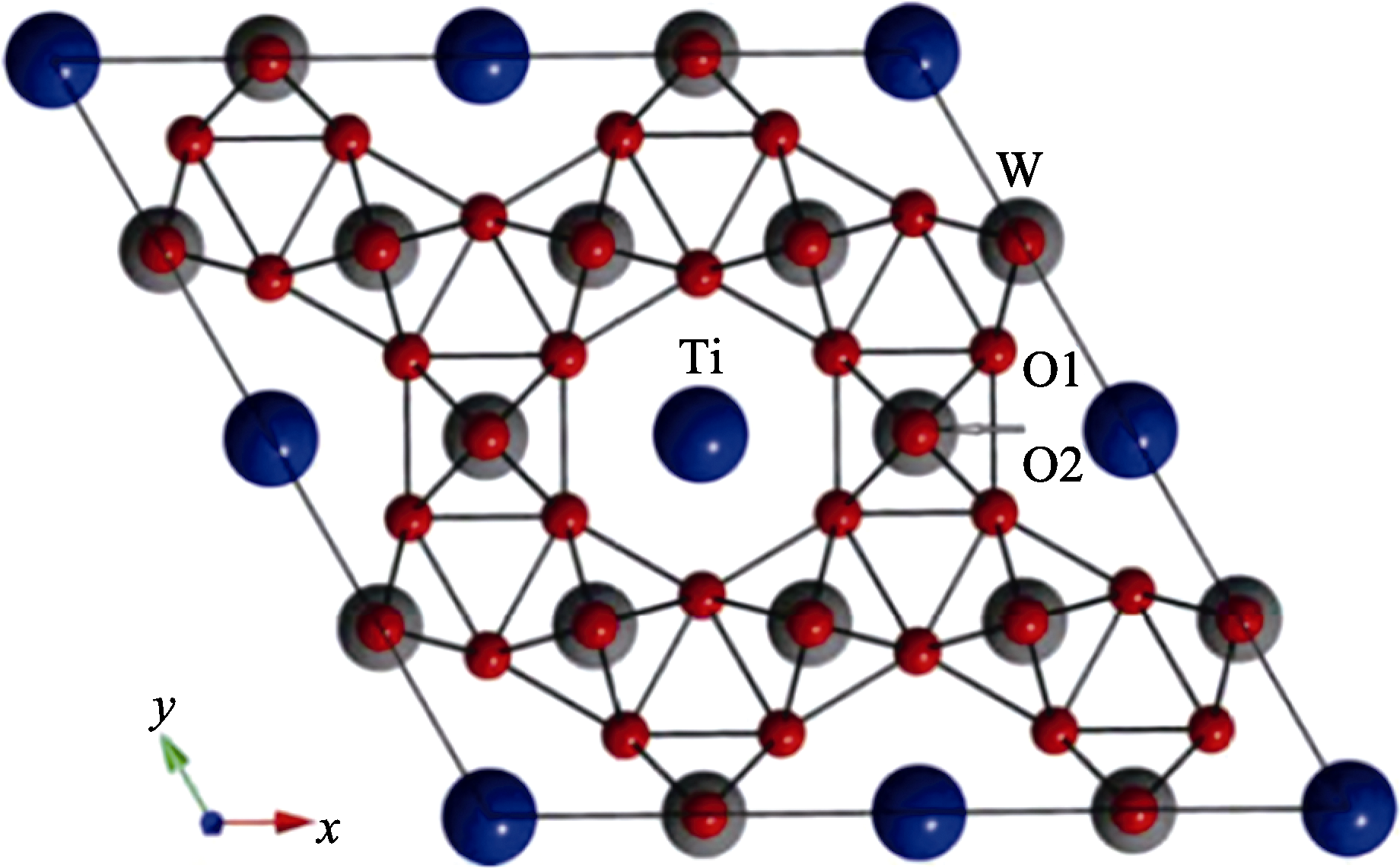
图5 Ti掺杂h-WO3的超晶胞俯视图[29]
Fig. 5 Top view of the supercell of Ti-doped h-WO3[29]
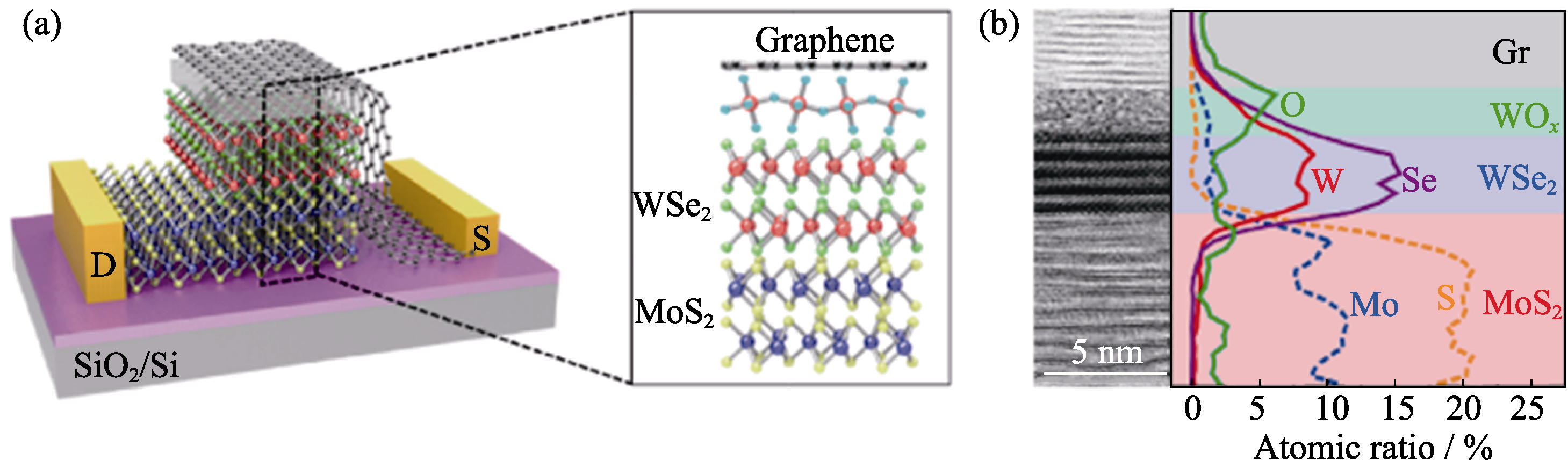
图6 WSe2-MoS2 p-n异质结(a)有氧化层的WOx/WSe2/MoS2设备截面示意图(粉球、蓝球、绿球、紫球、黄秋分别代表W、O、Se、Mo和S原子)和(b)截面的HR-TEM照片及EDS图[82]
Fig. 6 Monolithically band-engineered WSe2-MoS2 p-n heterojunction[82] (a) Schematic illustration of the vertical WSe2-MoS2 device with the WOx layer, where pink, blue, green, purple and yellow balls represent W, O, Se, Mo and S, respectively; (b) Cross-sectional HR-TEM image and EDS elemental line profiles across the WOx/WSe2/MoS2 heterointerfaces
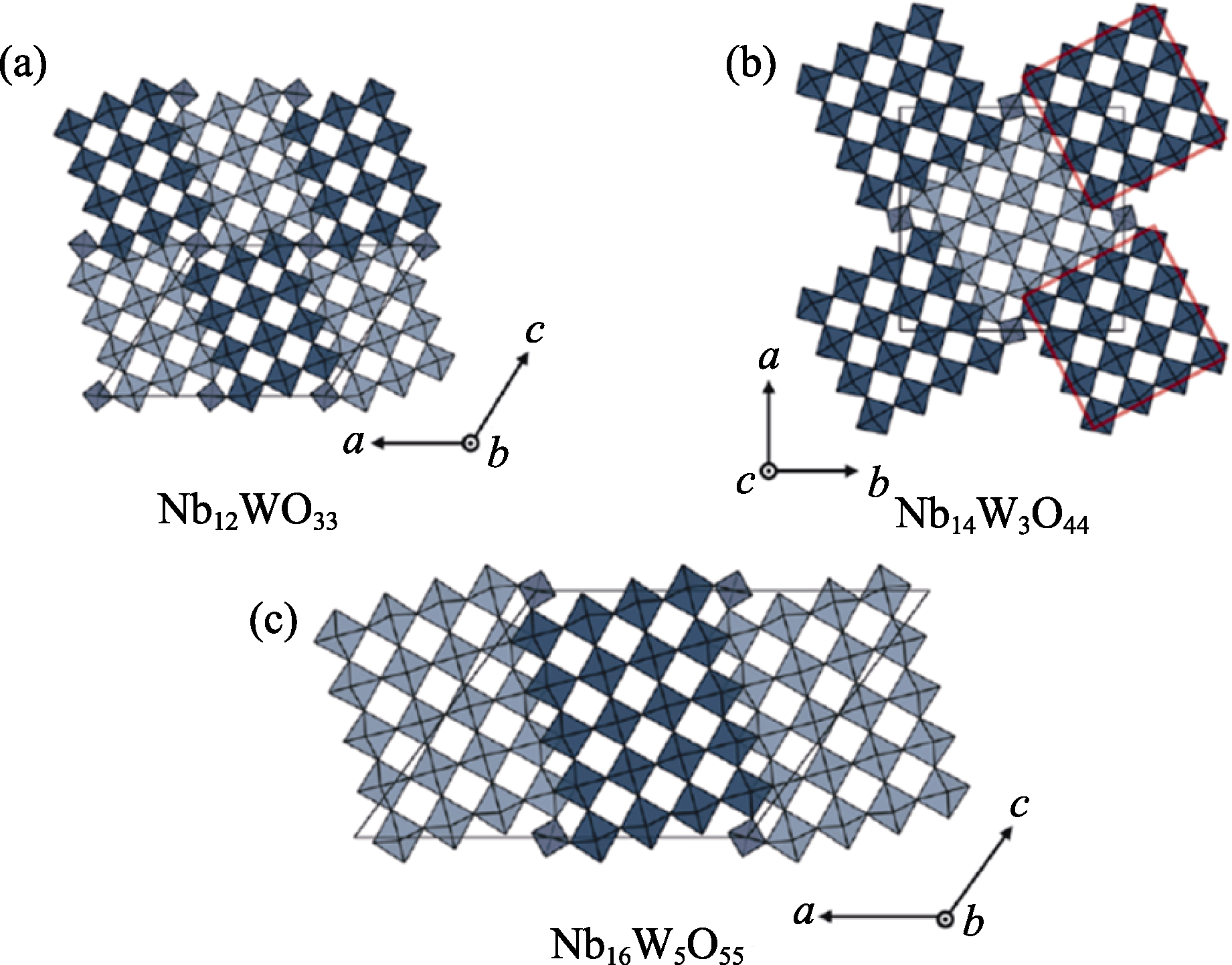
图7 Wadsley-Roth相晶体结构[85]
Fig. 7 Crystal structures of Wadsley-Roth phases[85] (a) Nb12WO33 (space group C2); (b) Nb14W3O44 (I4/m); (c) Nb16W5O55 (C2)
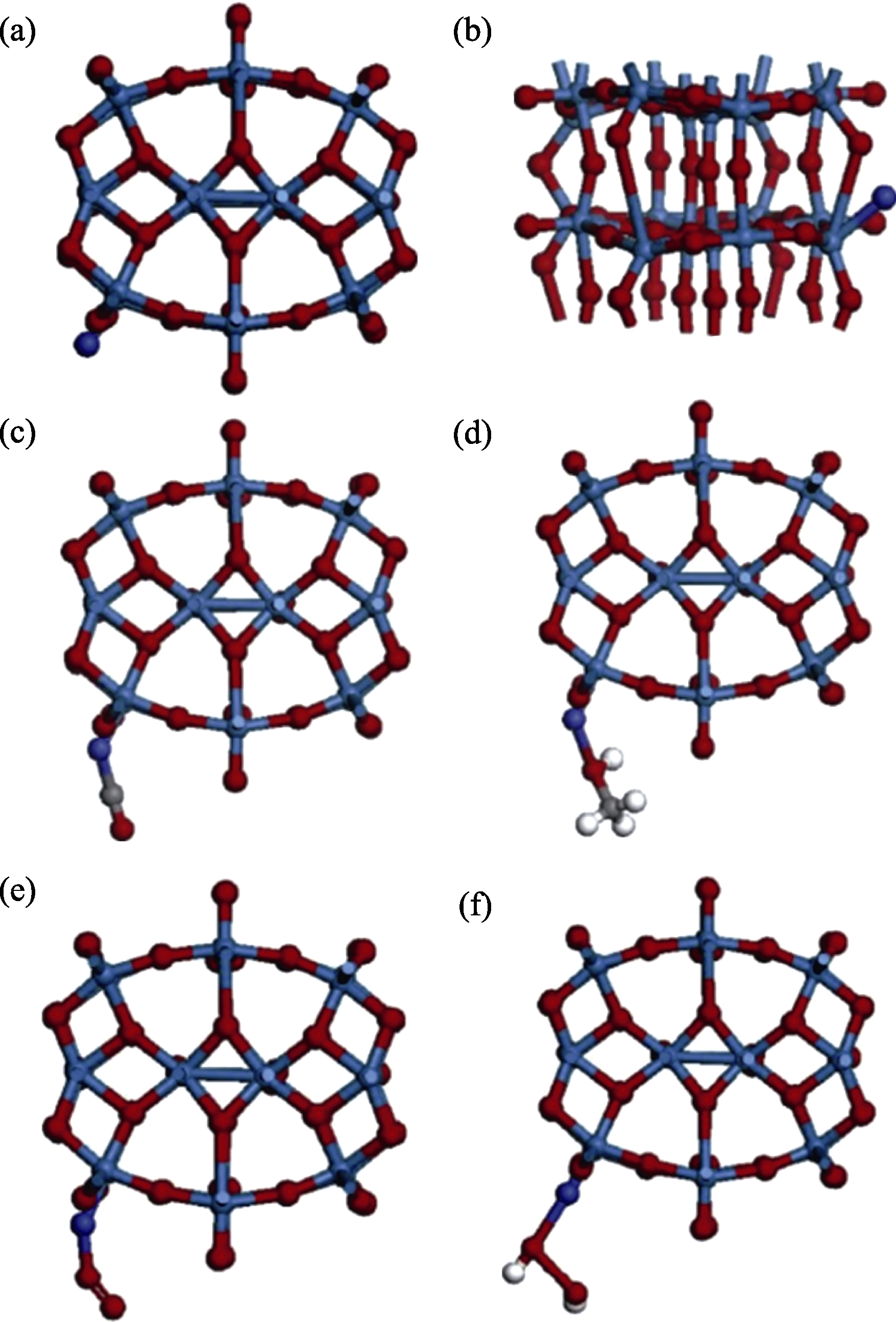
图8 优化后的不同气体分子在W18O49上的吸附模型(蓝球、紫球、红球、灰球、白球分别代表W、Co、O、C和H原子)[86]
Fig. 8 Optimized sadsorption model of different gas molecules on W18O49 with blue, purple, red, gray and white balls represent W, Co, O, C and H, respectively[86] (a, b) Adsorbed cobalt atom on the tungsten atom of NW (NW-Co); (c) Carbon monoxide molecule adsorbed on the NW-Co; (d) Methanol molecule adsorbed on the NW-Co; (e) Oxygen molecule adsorbed on the NW-Co; (f) Hydrogen peroxide molecule adsorbed on the NW-Co
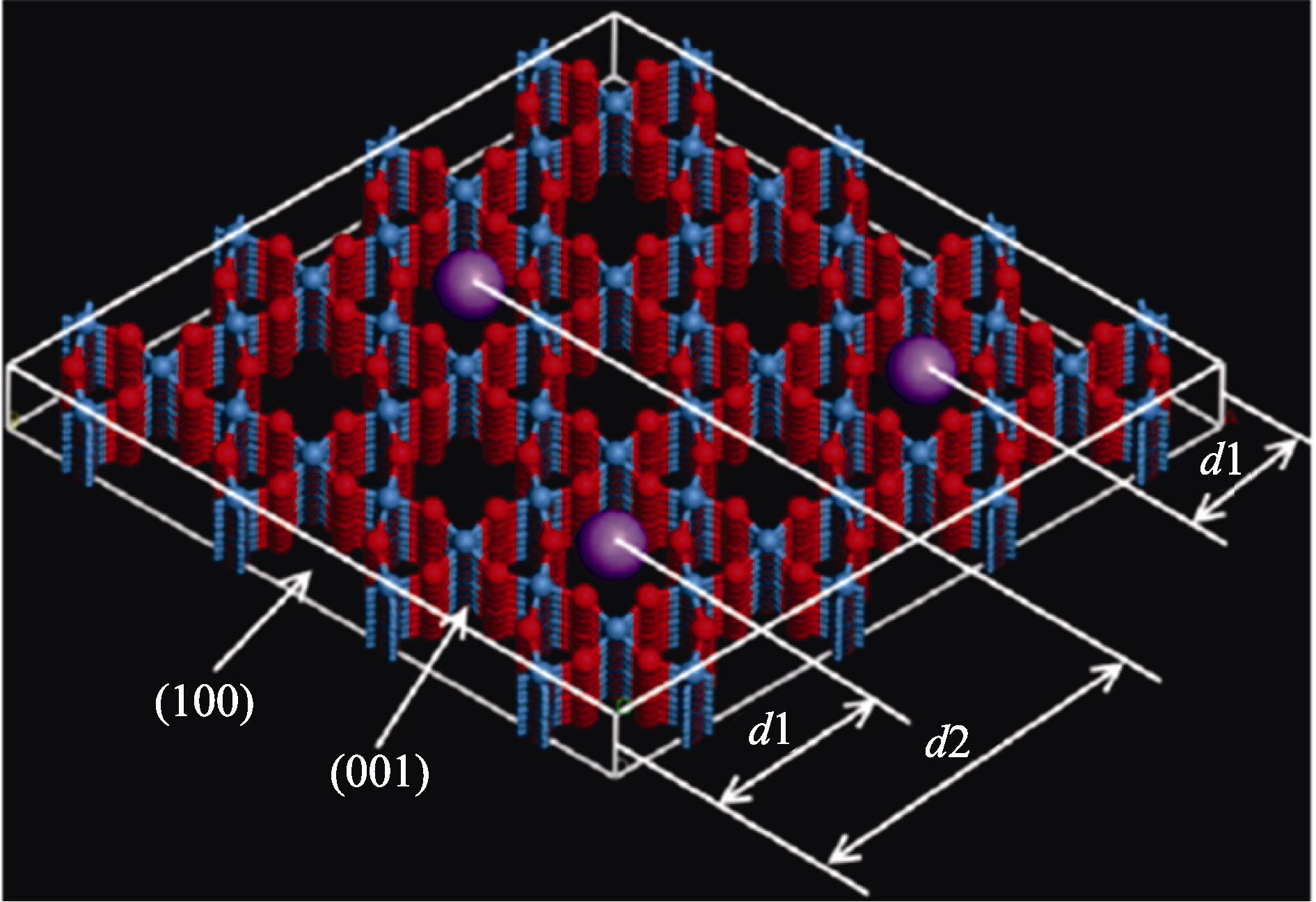
图9 h-WO3(100)面不同的嵌入位点示意图(蓝球、红球、紫球分别代表W、O原子和阳离子)[90]
Fig. 9 Various intercalating sites corresponding to different distances to the h-WO3(100) surface with blue, red and purple balls representing W, O and cations, respectively[90]
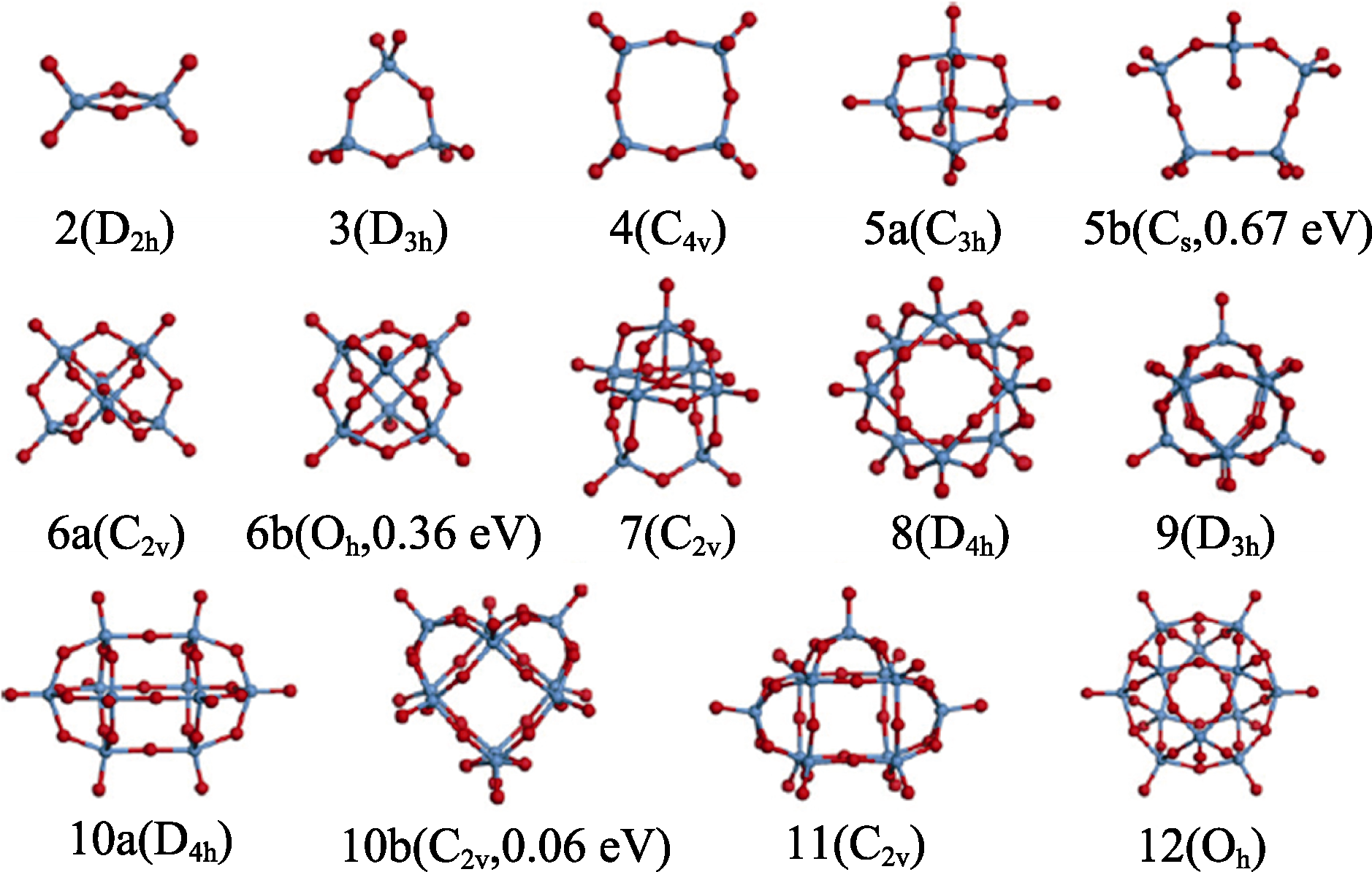
图10 (WO3)n簇(n=2~12)最低能量结构及其亚稳态同分异构体(5b、 6b、10b), 其中蓝球、红球分别代表W和O原子[95]
Fig. 10 Lowest-energy structures of (WO3)n clusters (n=2-12) and several metastable isomers (labeled as 5b, 6b, 10b) with blue and red balls representing W and O, respectively[95]
| [1] |
YIN X T, LV P, LI J, et al. Nanostructured tungsten trioxide prepared at various growth temperatures for sensing applications. Journal of Alloys and Compounds, 2020, 825:154105.
DOI URL |
| [2] |
NANDIYANTO A B D, OKTIANI R, RAGADHITA R, et al. Amorphous content on the photocatalytic performance of micrometer-sized tungsten trioxide particles. Arabian Journal of Chemistry, 2020, 13(1):2912-2924.
DOI URL |
| [3] |
HE X, WANG X Y, SUN B N, et al. Synthesis of three- dimensional hierarchical furball-like tungsten trioxide microspheres for high performance supercapacitor electrodes. RSC Advances, 2020, 10(23):13437-13441.
DOI URL |
| [4] |
HAI G J, HUANG J F, CAO L Y, et al. Influence of oxygen deficiency on the synthesis of tungsten oxide and the photocatalytic activity for the removal of organic dye. Journal of Alloys and Compounds, 2017, 690:239-248.
DOI URL |
| [5] |
LIU X F, ZHOU H, PEI S Z, et al. Oxygen-deficient WO3-x nanoplate array film photoanode for efficient photoelectrocatalytic water decontamination. Chemical Engineering Journal, 2020, 381:122740.
DOI URL |
| [6] | 赵林艳, 席晓丽, 樊佑书, 等. 纳米氧化钨的水热/溶剂热法制备及应用的综述. 材料导报, 2019, 33(19):3203-3209. |
| [7] |
QUAN H Q, GAO Y F, WANG W Z. Tungsten oxide-based visible light-driven photocatalysts: crystal and electronic structures and strategies for photocatalytic efficiency enhancement. Inorganic Chemistry Frontiers, 2020, 7(4):817-838.
DOI URL |
| [8] | PERSSON K. Materials project. https://materialsproject.org/ . |
| [9] |
DEB S K. Opportunities and challenges in science and technology of WO3 for electrochromic and related applications. Solar Energy Materials and Solar Cells, 2008, 92(2):245-258.
DOI URL |
| [10] |
DING Y, YANG I S, LI Z Q, et al. Nanoporous TiO2 spheres with tailored textural properties: controllable synthesis, formation mechanism, and photochemical applications. Progress in Materials Science, 2020, 109:100620.
DOI URL |
| [11] |
DONG P Y, HOU G H, XI X U, et al. WO3-based photocatalysts: morphology control, activity enhancement and multifunctional applications. Environmental Science-Nano, 2017, 4(3):539-557.
DOI URL |
| [12] |
HAN L F, CHEN J L, ZHANG Y H, et al. Facile synthesis of hierarchical carpet-like WO3 microflowers for high NO2 gas sensing performance. Materials Letters, 2018, 210:8-11.
DOI URL |
| [13] |
LI Y S, TANG Z L, ZHANG J Y, et al. Fabrication of vertical orthorhombic/hexagonal tungsten oxide phase junction with high photocatalytic performance. Applied Catalysis B-Environmental, 2017, 207:207-217.
DOI URL |
| [14] |
HUNGE Y M, YADAV A A, MAHADIK M A, et al. A highly efficient visible-light responsive sprayed WO3/FTO photoanode for photoelectrocatalytic degradation of brilliant blue. Journal of the Taiwan Institute of Chemical Engineers, 2018, 85:273-281.
DOI URL |
| [15] | KARADENIZ S M, TATAR D, ERTUGRUL M, et al. Structural, optical and electrochromic properties of WO3 thin films prepared by chemical spray pyrolysis versus spin coating technique. Spectroscopy and Spectral Analysis, 2018, 38(9):2982-2988. |
| [16] |
INAMDAR A I, CHAVAN H S, AHMED A A, et al. Nanograin tungsten oxide with excess oxygen as a highly reversible anode material for high-performance Li-ion batteries. Materials Letters, 2018, 215:233-237.
DOI URL |
| [17] |
SHENG J P, ZHANG L, DENG L, et al. Fabrication of dopamine enveloped WO3-x quantum dots as single-NIR laser activated photonic nanodrug for synergistic photothermal/photodynamic therapy against cancer. Chemical Engineering Journal, 2020, 383:123071.
DOI URL |
| [18] |
ZHAO L Y, XI X L, LIU Y S, et al. Growth mechanism and visible-light-driven photocatalysis of organic solvent dependent WO3 and nonstoichiometric WO3-x nanostructures. Journal of the Taiwan Institute of Chemical Engineers, 2020, 115:339-347.
DOI URL |
| [19] |
ZHAO L Y, XI X L, LIU Y S, et al. Facile synthesis of WO3 micro/nanostructures by paper-assisted calcination for visible- light-driven photocatalysis. Chemical Physics, 2020, 528:110515.
DOI URL |
| [20] |
FAN Y S, XI X L, LIU Y S, et al. Growth mechanism of immobilized WO3 nanostructures in different solvents and their visible-light photocatalytic performance. Journal of Physics and Chemistry of Solids, 2020, 140:109380.
DOI URL |
| [21] |
MANTHIRAM K, ALIVISATOS A P. Tunable localized surface plasmon resonances in tungsten oxide nanocrystals. Journal of the American Chemical Society, 2012, 134(9):3995-3998.
DOI URL |
| [22] |
KIMURA Y, IBANO K, UEHATA K, et al. Improved hydrogen gas sensing performance of WO3 films with fibrous nanostructured surface. Applied Surface Science, 2020, 532:147274.
DOI URL |
| [23] |
LI D, HUANG W Q, XIE Z, et al. Mechanism of enhanced photocatalytic activities on tungsten trioxide doped with sulfur: dopant-type effects. Modern Physics Letters B, 2016, 30(27):1650340.
DOI URL |
| [24] |
QIN Y X, LIU M, YE Z H. A DFT study on WO3 nanowires with different orientations for NO2 sensing application. Journal of Molecular Structure, 2014, 1076:546-553.
DOI URL |
| [25] |
CHEN Z J, CAO J X, YANG L W, et al. The unique photocatalysis properties of a 2D vertical MoO2 /WO2 heterostructure: a first- principles study. Journal of Physics D-Applied Physics, 2018, 51(26):265106.
DOI URL |
| [26] | 张秋杰, 高占忠, 原玉, 等. 第一性原理研究荷电状态对Pd13团簇催化分解NO性能的影响. 原子与分子物理学报, 2016, 33(03):438-442. |
| [27] |
JIA Q Q, JI H M, BAI X. Selective sensing property of triclinic WO3 nanosheets towards ultra-low concentration of acetone. Journal of Materials Science-Materials in Electronics, 2019, 30(8):7824-7833.
DOI URL |
| [28] |
MA Y L, FENG B, LANG J Y, et al. Synthesis of semimetallic tungsten trioxide for infrared light photoelectrocatalytic water splitting. Journal of Physical Chemistry C, 2019, 123(42):25833-25843.
DOI URL |
| [29] | 秦京运, 舒群威, 袁艺, 等. Ti0.33WO3电子结构和太阳辐射屏蔽性能第一性原理研究. 物理学报, 2020, 69(4):217-223. |
| [30] | DIRAC P A M. The Principles of Quantum Mechanics. Oxford: Clarendon Press, 1958: 1-22. |
| [31] | 曾瑾言. 量子力学. 北京, 科学出版社, 2000: 1-24. |
| [32] | BORN M, HUANG K. Dynamical Theory of Crystal Lattices. Oxford: Oxford University Press, 1958: 104-113. |
| [33] |
SLATER J C. Magnetic effects and the Hartree-Fock equation. Physical Review, 1951, 82(4):538-541.
DOI URL |
| [34] | 张跃, 谷景华, 商家香, 等. 计算材料学基础. 北京: 北京航天航空大学出版社, 2007: 83-135. |
| [35] | 徐光宪, 黎乐民, 王德民. 量子化学:基本原理和从头计算法. 北京: 科学出版社, 2007: 65-97. |
| [36] |
KOCH W, HOLTHAUSEN M C. A chemist's guide to density functional theory. Zeitschrift für Physik B Condensed Matter, 2001, 78(2):317-323.
DOI URL |
| [37] | KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects. Physical Review, 1965, 140:1133-1138. |
| [38] |
THOMAS H. The calculation of atomic fields. Proceedings of the Cambridge Philosophical Society, 1927, 23:542-548.
DOI URL |
| [39] |
KOHN W. Nobel lecture: electronic structure of matter-wave functions and density functionals. Reviews of Modern Physics, 1999, 71(5):1253-1266.
DOI URL |
| [40] | FERMI E. Un metodo statistico per la determinazione di alcune Priorieta dell atome. Rend. Accad. Naz. Lincei, 1927, 6:602. |
| [41] | DIRAC P A M. Note on exchange phenomena in the thomas-fermi atom. Proceedings of the Cambridge Philosophical Royal Society, 1930, 26:376. |
| [42] | SLATER J C. A simplification of the hartree-fock method. Self-Consistent Fields in Atoms, 1975, 81(3):215-230. |
| [43] |
PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, molecules, solids, and surfaces-applications of the generalized gradient approximation for exchange and correlation. Physical Review B, 1992, 46(11):6671-6687.
DOI URL |
| [44] | 谢希德, 陆栋. 固体能带理论. 上海: 复旦大学出版社, 1998: 66-69. |
| [45] |
VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Physical Review B, 1990, 41(11):7892-7895.
DOI URL |
| [46] |
BLÖCHL P E. Projector augmented-wave method. Physical Review B Condens Matter, 1994, 50(24):17953-17959.
DOI URL |
| [47] |
HAMANN D R, SCHLÜTER M, CHIANG C. Norm-conserving pseudopotentials. Physical Review Letters, 1979, 43(20):1494-1497.
DOI URL |
| [48] | 北京创腾科技有限公司. BIOVIA materials studio.[EB/OL]. [2021-04-26] https://www.neotrident.com/index.php/product/proinfo/29.html . |
| [49] | 北京宏剑公司. Vienna ab-initio simulation package. [EB/OL]. [2021-04-26] http://www.hongcam.com.cn/index.php/product/materials/vasp . |
| [50] | WEB V. Psi-k-Ab initio. [EB/OL]. [2021-04-26] http://psi-k.net/software/ . |
| [51] | KAMINSKY W. WinXMorph. [EB/OL]. [2021-04-26] http://cad4.cpac.washington.edu/WinXMorphHome/WinXMorph.htm#opennewwindow . |
| [52] | MOMMA K. Visualization for electronic and structural analysis. [EB/OL]. (2021-01-09) [2021-04-26] http://www.jp-minerals.org/vesta/en/ . |
| [53] |
MAHAJAN S, JAGTAP S. Metal-oxide semiconductors for carbon monoxide (CO) gas sensing: a review. Applied Materials Today, 2020, 18:100483.
DOI URL |
| [54] |
ZEB S, PENG X J, YUAN G Z, et al. Controllable synthesis of ultrathin WO3 nanotubes and nanowires with excellent gas sensing performance. Sensors and Actuators B-Chemical, 2020, 305:127435.
DOI URL |
| [55] |
LIU D, REN X W, LI Y S, et al. Nanowires-assembled WO3 nanomesh for fast detection of ppb-level NO2 at low temperature. Journal of Advanced Ceramics, 2020, 9(2):17-26.
DOI URL |
| [56] |
OISON V, SAADI L, LAMBERT-MAURIAT C, et al. Mechanism of CO and O3 sensing on WO3 surfaces: first principle study. Sensors and Actuators B-Chemical, 2011, 160(1):505-510.
DOI URL |
| [57] |
ZHAO L H, TIAN F H, WANG X B, et al. Mechanism of CO adsorption on hexagonal WO3(001) surface for gas sensing: a DFT study. Computational Materials Science, 2013, 79:691-697.
DOI URL |
| [58] |
JIN H, ZHOU H, ZHANG Y F. Insight into the mechanism of CO oxidation on WO3(001) surfaces for gas sensing: a DFT study. Sensors, 2017, 17(8):1898.
DOI URL |
| [59] | TANG B L, JIANG G H, CHEN W X, et al. First-principles study on hexagonal WO3 for HCHO gas sensing application. Acta Metallurgica Sinica-English Letters, 2015, 28(6):772-780. |
| [60] | HAN X, YIN X H. Density functional theory study of the NO2-sensing mechanism on a WO3(001) surface: the role of surface oxygen vacancies in the formation of NO and NO3. Molecular Physics, 2016, 114(24):3546-3555. |
| [61] | QIN Y X, LIU M, HUA D Y. First-principles study of the electronic structure and NO2-sensing properties of Ti-doped W18O49 nanowire. Acta Physica Sinica, 2014, 63(20):207101. |
| [62] | QIN Y X, YE Z H. DFT study on interaction of NO2 with the vacancy-defected WO3 nanowires for gas-sensing. Sensors and Actuators B-Chemical, 2016, 222:499-507. |
| [63] | BAI S L, ZHANG K W, WANG L S, et al. Synthesis mechanism and gas-sensing application of nanosheet-assembled tungsten oxide microspheres. Journal of Materials Chemistry A, 2014, 2(21):7927-7934. |
| [64] | YAKOVKIN I N, GUTOWSKI M. Driving force for the WO3(001) surface relaxation. Surface Science, 2007, 601(6):1481-1488. |
| [65] | 张克伟. 气敏和光催化导向的氧化钨纳米结构设计与改性. 北京: 北京化工大学博士学位论文, 2014. |
| [66] | YANG H H, SUN H G, LI Q T, et al. Structural, electronic, optical and lattice dynamic properties of the different WO3 phases: first-principle calculation. Vacuum, 2019, 164:411-420. |
| [67] | 杨欢欢. 第一性原理研究WO3低指数表面上H2O分子吸附和分解的微观机制. 济南: 山东大学硕士学位论文, 2019. |
| [68] | ZHENG T T, SANG W, HE Z H, et al. Conductive tungsten oxide nanosheets for highly efficient hydrogen evolution. Nano Letters, 2017, 17(12):7968-7973. |
| [69] | 桑炜. 纳米晶可控合成以及催化性能的调控. 合肥: 中国科学技术大学博士学位论文, 2018. |
| [70] | WANG F G, DI VALENTIN C, PACCHIONI G. Doping of WO3 for photocatalytic water splitting: hints from density functional theory. Journal of Physical Chemistry C, 2012, 116(16):8901-8909. |
| [71] | ZHANG T, ZHU Z L, CHEN H N, et al. Iron-doping-enhanced photoelectrochemical water splitting performance of nanostructured WO3: a combined experimental and theoretical study. Nanoscale, 2015, 7(33):2933-2940. |
| [72] | HUANG W C, WANG J X, BIAN L, et al. Oxygen vacancy induces self-doping effect and metalloid LSPR in non-stoichiometric tungsten suboxide synergistically contributing to the enhanced photoelectrocatalytic performance of WO3-x/TiO2-x heterojunction. Physical Chemistry Chemical Physics, 2018, 20(25):17268-17278. |
| [73] | ZHANG N, LI X Y, LIU Y F, et al. Defective tungsten oxide hydrate nanosheets for boosting aerobic coupling of amines: synergistic catalysis by oxygen vacancies and bronsted acid sites. Small, 2017, 13(31):1701354. |
| [74] | ZHANG N, JALIL A, WU D X, et al. Refining defect states in W18O49 by Mo doping: a strategy for tuning N2 activation towards solar-driven nitrogen fixation. Journal of the American Chemical Society, 2018, 140(30):9434-9443. |
| [75] | ZHANG N, LONG R, GAO C, et al. Recent progress on advanced design for photoelectrochemical reduction of CO2 to fuels. Science China-Materials, 2018, 61(6):771-805. |
| [76] | LI M Q, ZHANG N, LONG R, et al. PdPt alloy nanocatalysts supported on TiO2: maneuvering metal-hydrogen interactions for light-driven and water-donating selective alkyne semihydrogenation. Small, 2017, 13(23):1604173. |
| [77] | WANG Z, WANG X Y, CONG S, et al. Fusing electrochromic technology with other advanced technologies: a new roadmap for future development. Materials Science & Engineering R-Reports, 2020, 140:100524. |
| [78] | YAO Y J, ZHAO Q, WEI W, et al. WO3 quantum-dots electrochromism. Nano Energy, 2020, 68:104350. |
| [79] | LIN H, ZHOU F, LIU C P, et al. Non-grotthuss proton diffusion mechanism in tungsten oxide dihydrate from first-principles calculations. Journal of Materials Chemistry A, 2014, 2(31):12280-12288. |
| [80] | HJELM A, GRANQVIST C G, WILLS J M. Electronic structure and optical properties of WO3, LiWO3, NaWO3, and HWO3. Physical Review B, 1996, 54(4):2436-2445. |
| [81] | WISEMAN P J, DICKENS P G. Neutron-diffraction studies of cubic tungsten bronzes. Journal of Solid State Chemistry, 1976, 17(1/2):91-100. |
| [82] | YANG S, CHA J, KIM J C, et al. Monolithic interface contact engineering to boost optoelectronic performances of 2D semiconductor photovoltaic heterojunctions. Nano Letters, 2020, 20(4):2443-2451. |
| [83] | 方城, 汪洪, 施思齐. 氧化钨电致变色性能的研究进展. 物理学报, 2016, 65(16):168201. |
| [84] | 高占忠. 锂离子电池负极材料WO3的第一性原理研究. 成都: 电子科技大学硕士学位论文, 2017. |
| [85] | KOCER C P, GRIFFITH K J, GREY C P, et al. Cation disorder and lithium insertion mechanism of Wadsley-Roth crystallographic shear phases from first principles. Journal of the American Chemical Society, 2019, 141(38):15121-15134. |
| [86] | KARIM N A, KAMARUDIN S K, SHYUAN L K, et al. Study on the electronic properties and molecule adsorption of W18O49 nanowires as a catalyst support in the cathodes of direct methanol fuel cells. Journal of Power Sources, 2015, 288:461-472. |
| [87] | ZHANG Z F, CHEN J L, LI H B, et al. Vapor-solid nanotube growth via sidewall epitaxy in an environmental transmission electron microscope. Crystal Growth & Design, 2017, 17(1):11-15. |
| [88] | ZHANG Z F, WANG Y, LI H B, et al. Atomic-scale observation of vapor-solid nanowire growth via oscillatory mass transport. ACS Nano, 2016, 10(1):763-769. |
| [89] | ZHANG Z F, SHENG L P, CHEN L, et al. Atomic-scale observation of pressure-dependent reduction dynamics of W18O49 nanowires using environmental TEM. Physical Chemistry Chemical Physics, 2017, 19(25):16307-16311. |
| [90] | CHEN L, LAM S, ZENG Q H, et al. Effect of cation intercalation on the growth of hexagonal WO3 nanorods. Journal of Physical Chemistry C, 2012, 116(21):11722-11727. |
| [91] | JIANG S, CHEKINI M, QU Z B, et al. Chiral ceramic nanoparticles and peptide catalysis. Journal of the American Chemical Society, 2017, 139(39):13701-13712. |
| [92] | GU L J, MA C L, ZHANG X H, et al. Populating surface-trapped electrons towards SERS enhancement of W18O49 nanowires. Chemical Communications, 2018, 54(49):6332-6335. |
| [93] | MEHMOOD F, PACHTER R, MURPHY N R, et al. Effect of oxygen vacancies on the electronic and optical properties of tungsten oxide from first principles calculations. Journal of Applied Physics, 2016, 120(23):233105. |
| [94] | MIGAS D B, SHAPOSHNIKOV V L, RODIN V N, et al. Tungsten oxides. I. Effects of oxygen vacancies and doping on electronic and optical properties of different phases of WO3. Journal of Applied Physics, 2010, 108(9):093713. |
| [95] | SAI L W, TANG L L, HUANG X M, et al. Lowest-energy structures of (WO3)n(2≤n≤12) clusters from first-principles global search. Chemical Physics Letters, 2012, 544:7-12. |
| [96] | HUANG X, ZHAI H J, LI J, et al. On the structure and chemical bonding of tri-tungsten oxide clusters W3On- and W3On (n=7-10): W3O8 as a potential molecular model for O-deficient defect sites in tungsten oxides. Journal of Physical Chemistry A, 2006, 110(1):85-92. |
| [97] | 邱克强, 王爱民, 张海峰, 等. 钨丝增强ZrAlNiCuSi块体非晶复合材料及其塑性行为. 金属学报, 2002(10):1091-1096. |
| [98] | JIANG P G, XIAO Y Y, LIU W J, et al. Hydrogen reduction characteristics of WO3 based on density functional theory. Results in Physics, 2019, 12:896-902. |
| [99] | LIU W J, JIANG P G, XIAO Y Y, et al. A study of the hydrogen adsorption mechanism of W18O49 using first-principles calculations. Computational Materials Science, 2018, 154:53-59. |
| [100] | 宋翰林, 姜平国, 刘文杰, 等. 氧化钨氢还原动力学的研究进展. 有色金属科学与工程, 2017, 8(5):64-69. |
| [101] | 姜平国, 汪正兵, 闫永播. 三氧化钨表面氢吸附机理的第一性原理研究. 物理学报, 2017, 66(8):294-303. |
| [1] | 丁玲, 蒋瑞, 唐子龙, 杨运琼. MXene材料的纳米工程及其作为超级电容器电极材料的研究进展[J]. 无机材料学报, 2023, 38(6): 619-633. |
| [2] | 杨卓, 卢勇, 赵庆, 陈军. X射线衍射Rietveld精修及其在锂离子电池正极材料中的应用[J]. 无机材料学报, 2023, 38(6): 589-605. |
| [3] | 陈强, 白书欣, 叶益聪. 热管理用高导热碳化硅陶瓷基复合材料研究进展[J]. 无机材料学报, 2023, 38(6): 634-646. |
| [4] | 林俊良, 王占杰. 铁电超晶格的研究进展[J]. 无机材料学报, 2023, 38(6): 606-618. |
| [5] | 张守超, 陈洪雨, 刘洪飞, 杨羽, 李欣, 刘德峰. 6H-SiC中子辐照肿胀高温回复及光学特性研究[J]. 无机材料学报, 2023, 38(6): 678-686. |
| [6] | 杨颖康, 邵怡晴, 李柏良, 吕志伟, 王路路, 王亮君, 曹逊, 吴宇宁, 黄荣, 杨长. Cl掺杂对CuI薄膜发光性能增强研究[J]. 无机材料学报, 2023, 38(6): 687-692. |
| [7] | 牛嘉雪, 孙思, 柳鹏飞, 张晓东, 穆晓宇. 铜基纳米酶的特性及其生物医学应用[J]. 无机材料学报, 2023, 38(5): 489-502. |
| [8] | 苑景坤, 熊书锋, 陈张伟. 聚合物前驱体转化陶瓷增材制造技术研究趋势与挑战[J]. 无机材料学报, 2023, 38(5): 477-488. |
| [9] | 杜剑宇, 葛琛. 光电人工突触研究进展[J]. 无机材料学报, 2023, 38(4): 378-386. |
| [10] | 杨洋, 崔航源, 祝影, 万昌锦, 万青. 柔性神经形态晶体管研究进展[J]. 无机材料学报, 2023, 38(4): 367-377. |
| [11] | 游钧淇, 李策, 杨栋梁, 孙林锋. 氧化物双介质层忆阻器的设计及应用[J]. 无机材料学报, 2023, 38(4): 387-398. |
| [12] | 林思琪, 李艾燃, 付晨光, 李荣斌, 金敏. Zintl相Mg3X2(X=Sb, Bi)基晶体生长及热电性能研究进展[J]. 无机材料学报, 2023, 38(3): 270-279. |
| [13] | 陈昆峰, 胡乾宇, 刘锋, 薛冬峰. 多尺度晶体材料的原位表征技术与计算模拟研究进展[J]. 无机材料学报, 2023, 38(3): 256-269. |
| [14] | 张超逸, 唐慧丽, 李宪珂, 王庆国, 罗平, 吴锋, 张晨波, 薛艳艳, 徐军, 韩建峰, 逯占文. 新型GaN与ZnO衬底ScAlMgO4晶体的研究进展[J]. 无机材料学报, 2023, 38(3): 228-242. |
| [15] | 齐占国, 刘磊, 王守志, 王国栋, 俞娇仙, 王忠新, 段秀兰, 徐现刚, 张雷. GaN单晶的HVPE生长与掺杂进展[J]. 无机材料学报, 2023, 38(3): 243-255. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||