Journal of Inorganic Materials ›› 2022, Vol. 37 ›› Issue (5): 541-546.DOI: 10.15541/jim20210280
• RESEARCH ARTICLE • Previous Articles Next Articles
WANG Peng( ), JIN Zunlong(), CHEN Ningguang, LIU Yonghao
), JIN Zunlong(), CHEN Ningguang, LIU Yonghao
Received:2021-04-29
Revised:2021-06-07
Published:2022-05-20
Online:2021-05-20
Contact:
JIN Zunlong, professor. E-mail: zljin@zzu.edu.cn
About author:WANG Peng (1995-), male, Master candidate. E-mail: 770352508@qq.com
Supported by:CLC Number:
WANG Peng, JIN Zunlong, CHEN Ningguang, LIU Yonghao. Theoretical Investigation of Mo Doped α-MnO2 Electrocatalytic Oxygen Evolution Reaction[J]. Journal of Inorganic Materials, 2022, 37(5): 541-546.
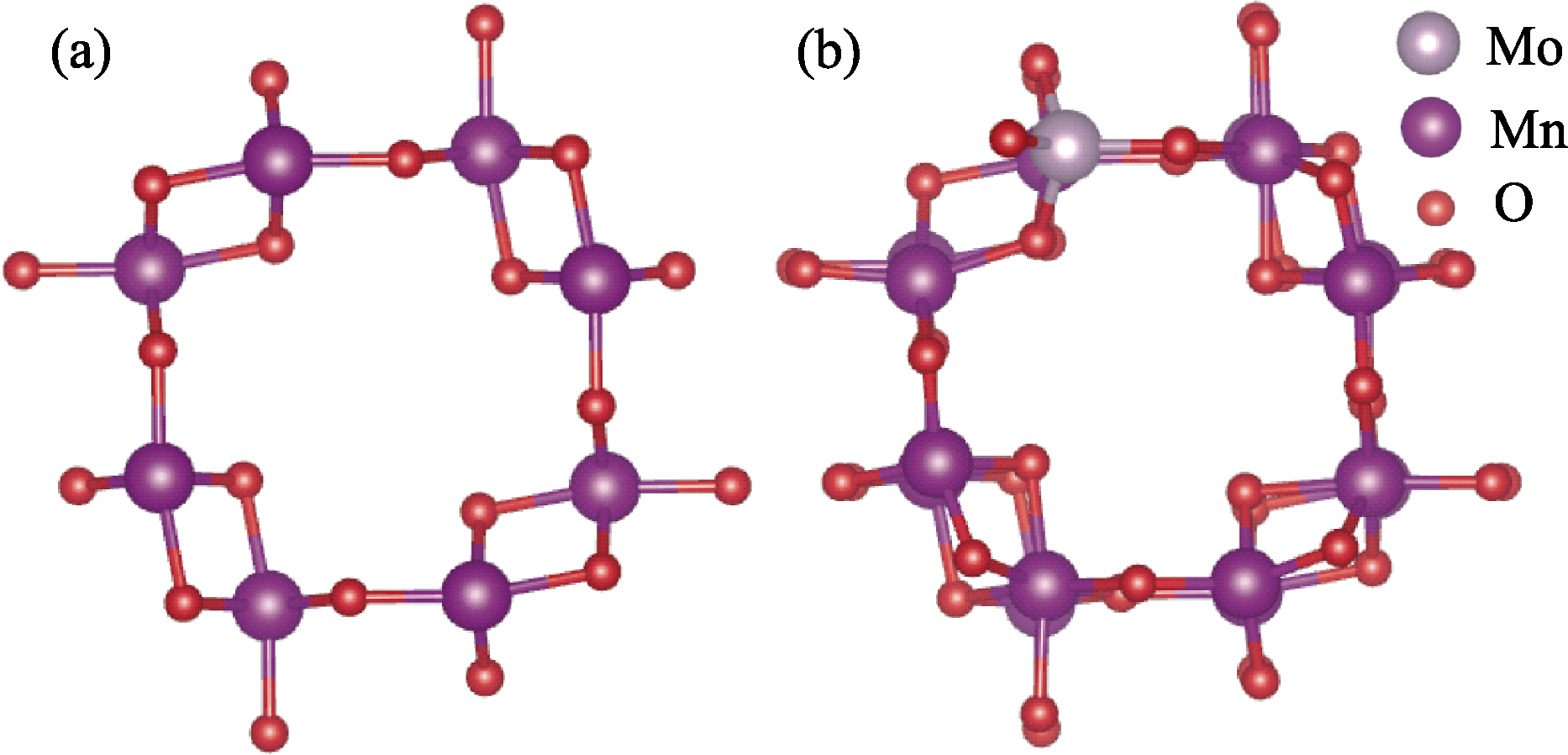
Fig. 1 Top view of optimized structures for (a) α-MnO2(001) and (b)Mo doped α-MnO2(001)
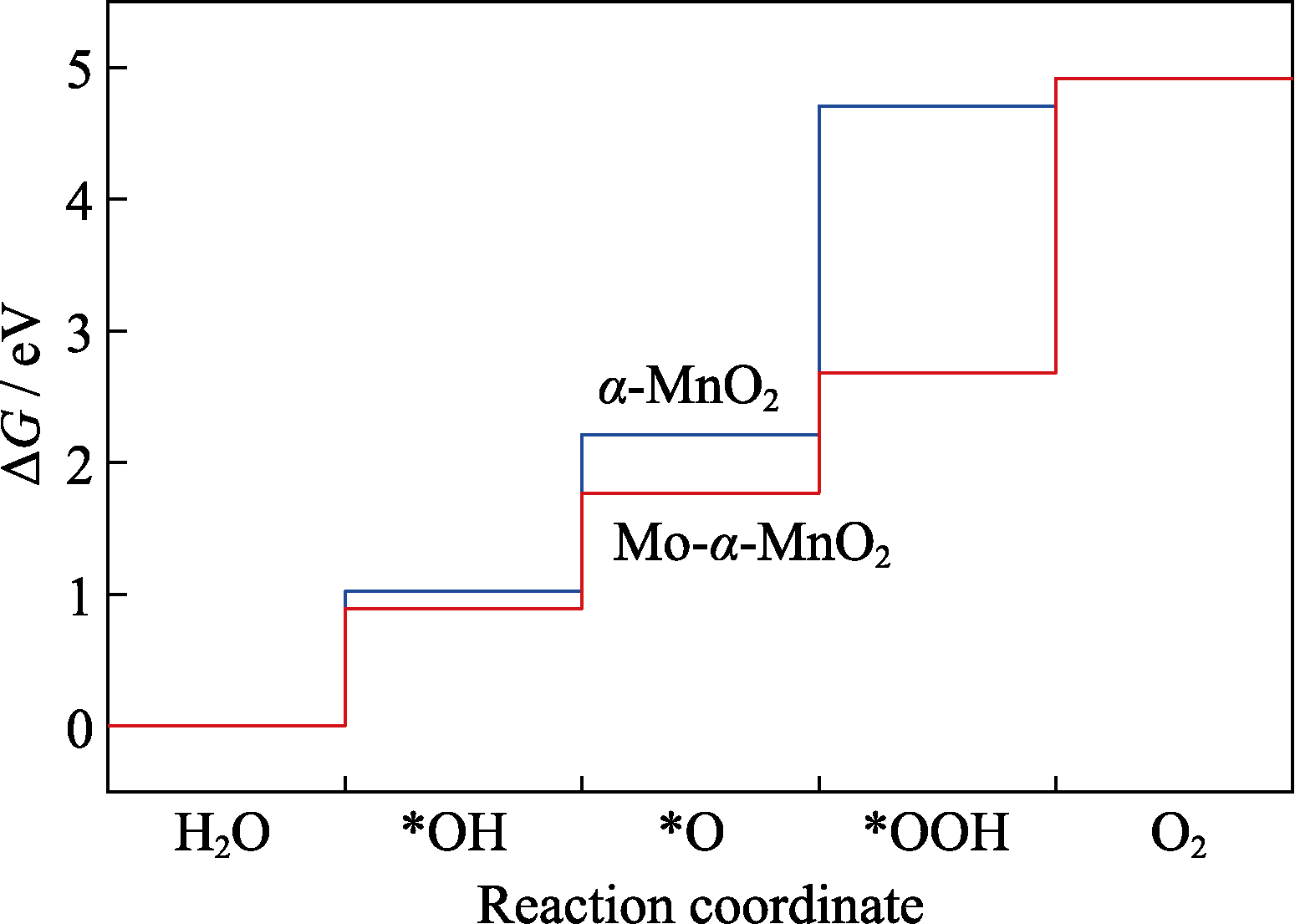
Fig. 2 OER free energy diagrams of α-MnO2 and Mo doped α-MnO2
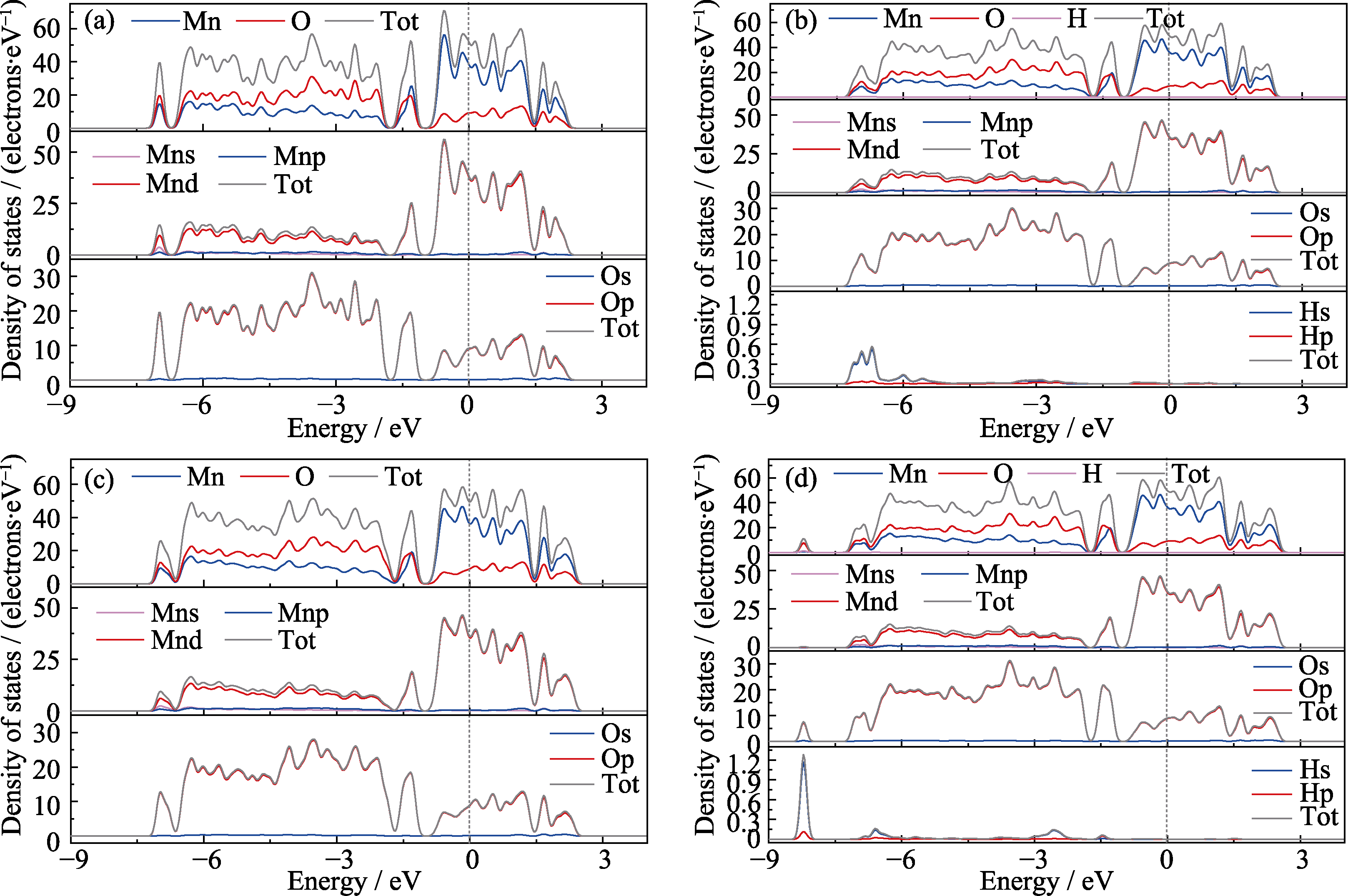
Fig. 3 DOS of α-MnO2 adsorbing different intermediates (a) Undoped; (b)*OH; (c)*O; (d)*OOH Colorful figures are available on website
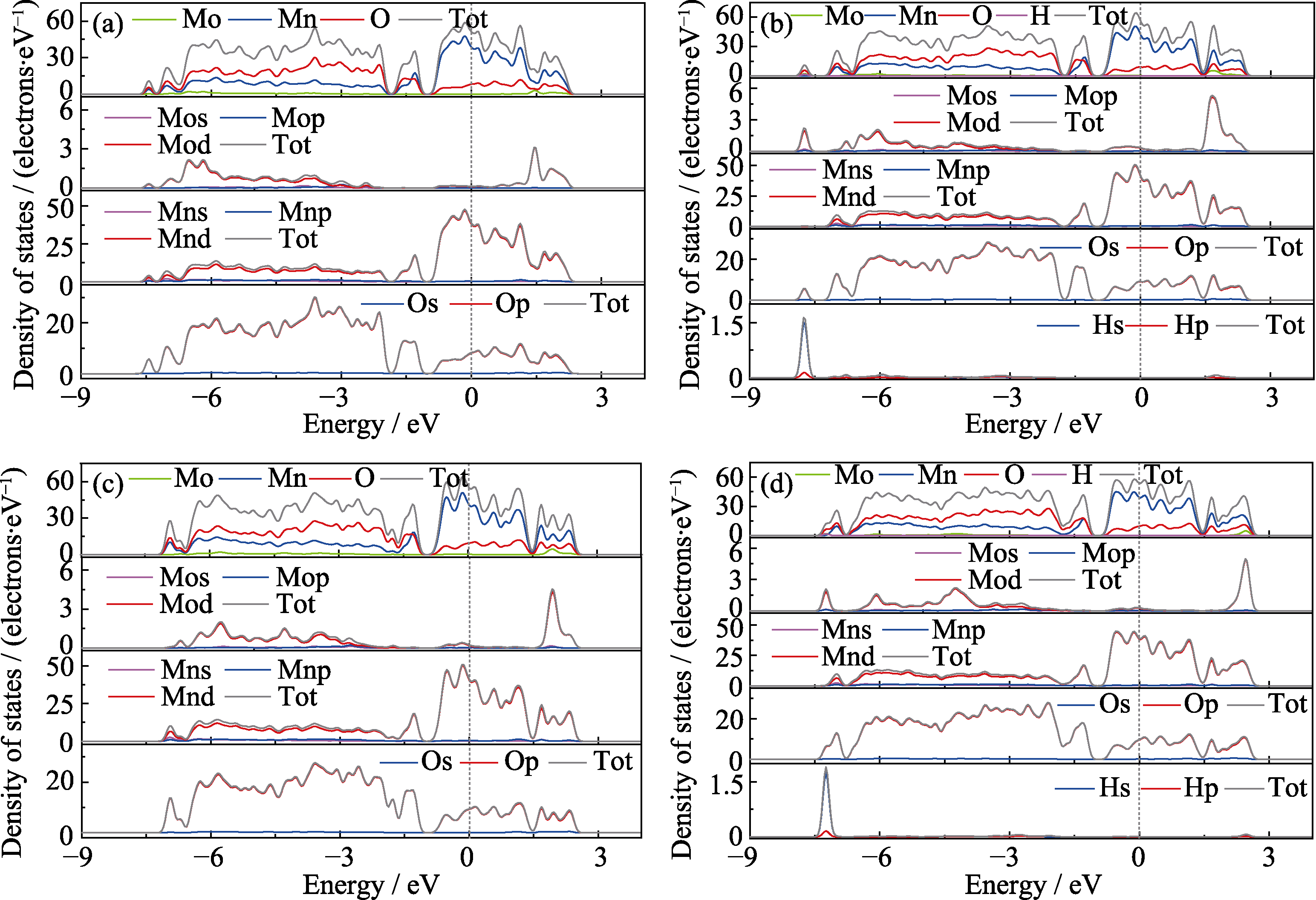
Fig. 4 DOS of Mo-doped α-MnO2 adsorbing different intermediates (a) Undoped; (b)*OH; (c)*O; (d)*OOH Colorful figures are available on website
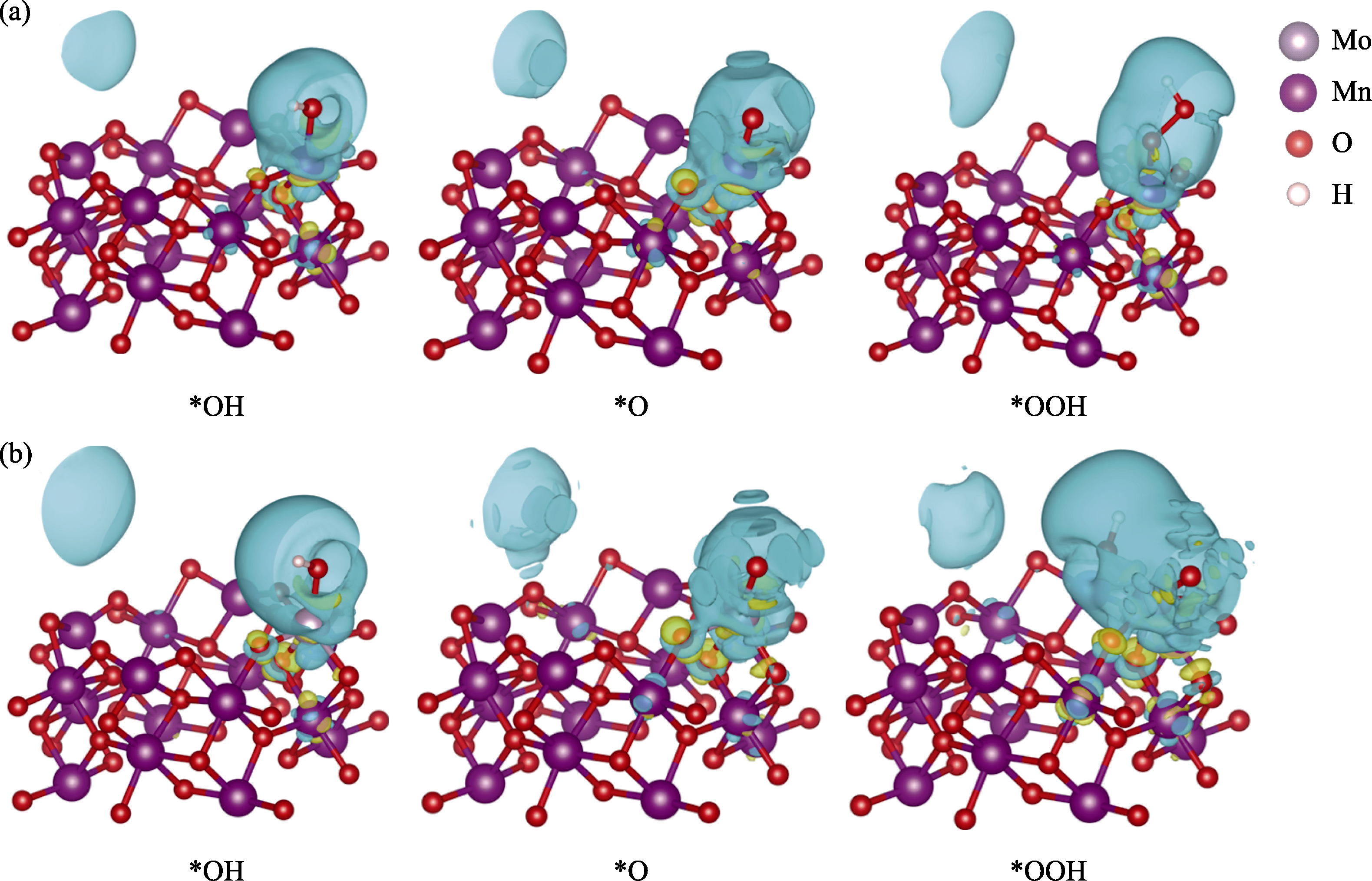
Fig. 5 Charge density difference of (a) α-MnO2 and (b) Mo doped α-MnO2 adsorbing different intermediates Blue area refers consuming charge; Yellow area refers assembling charge
| [1] | JAMESH M I. Recent progress on earth abundant hydrogen evolution reaction and oxygen evolution reaction bifunctional electrocatalyst for overall water splitting in alkaline media. Journal of Power Sources, 2016,333:213-236. |
| [2] | SHEN J Q, LIAO P Q, ZHOU D D, et al. Modular and stepwise synthesis of a hybrid metal-organic framework for efficient electrocatalytic oxygen evolution. Journal of the American Chemical Society, 2017,139:1778-1781. |
| [3] | ZHANG Y C, AFZAL N, PAN L, et al. Structure-activity relationship of defective metal-based photocatalysts for water splitting: experimental and theoretical perspectives. Advanced Science, 2019,6:1900053. |
| [4] | LIM J, PARK D, JEON S S, et al. Ultrathin IrO2 nanoneedles for electrochemical water oxidation. Advanced Functional Materials, 2018,28(4):1704796. |
| [5] | ZHAO Z H, ZhANG L P, XIA Z H, et al. Electron transfer and catalytic mechanism of organic molecule-adsorbed graphene nanoribbons as efficient catalysts for oxygen reduction and evolution reactions. Journal of Physical Chemistry C, 2016,120:2166-2175. |
| [6] | ZHAO Z H, XIA Z H. Design principles for dual-element-doped carbon nanomaterials as efficient bifunctional catalysts for oxygen reduction and evolution reactions. ACS Catalysis, 2016,6(3):1553-1558. |
| [7] | YE Z G, QIN C L, MA G, et al. Cobalt-iron oxide nanoarrays supported on carbon fiber paper with high stability for electrochemical oxygen evolution at large current densities. ACS Applied Materials & Interfaces, 2018,10(46):39809-39818. |
| [8] | ANANTHARAJ S, REDDY P N, KUNDU S. Core-oxidized amorphous cobalt phosphide nanostructures: an advanced and highly efficient oxygen evolution catalyst. Inorganic Chemistry, 2017,56(3):174-1756. |
| [9] | YU L, YANG J F, GUAN B Y, et al. Hierarchical hollow nanoprisms based on ultrathin Ni-Fe layered double hydroxide nanosheets with enhanced electrocatalytic activity towards oxygen evolution. Angewandte Chemie-International Edition, 2018,57(1):172-176. |
| [10] | BURKE M S, KAST M G, TROTOCHAUD L, et al. Cobalt-iron (oxy) hydroxide oxygen evolution electrocatalysts: the role of structure and composition on activity, stability, and mechanism. Journal of the American Chemical Society, 2015,137(10):3638-3648. |
| [11] | TROTOCHAUD L, YOUNG S L, RANNEY J K, et al. Nickel- iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. Journal of the American Chemical Society, 2014,136:6744-6753. |
| [12] | PARGOLETTI E, CAPPELLETTI G, MINGUZZI A, et al. High- performance of bare and Ti-doped α-MnO2 nanoparticles in catalyzing the oxygen reduction reaction. Journal of Power Sources, 2016,325:116-128. |
| [13] | ZHAO Y F, ZHANG J Q, WUA W J, et al. Cobalt-doped MnO2 ultrathin nanosheets with abundant oxygen vacancies supported on functionalized carbon nanofibers for efficient oxygen evolution. Nano Energy, 2018,54:129-137. |
| [14] | LEHTIMAEKI M, HOFFMANNOVA H, BOYTSOVA O, et al. Targeted design of α-MnO2 based catalysts for oxygen reduction. Electrochimica Acta, 2016,191:452-461. |
| [15] | SELVAKUMAR K, KUMAR S M S, THANGAMUTHU R, et al. Development of shape-engineered a-MnO2 materials as bi-functional catalysts for oxygen evolution reaction and oxygen reduction reaction in alkaline medium. International Journal of Hydrogen Energy, 2014,39(36):21024-21036. |
| [16] | XIA W, MAHMOOD A, LIANG Z B, et al. Earth-abundant nanomaterials for oxygen reduction. Angewandte Chemie- International Edition, 2016,55(8):2650-2676. |
| [17] | KRESSE G, HAFNER J. Ab initio molecular-dynamics simulation of the liquid etalamorphous-semiconductor transition in germanium. Physical Review B, 1994,49:14251-14269. |
| [18] | BLOCHL P E. Projector augmented-wave method. Phys. Rev. B, 1994,50(24):17953-17979. |
| [19] | PERDEW J P, BURKE K, ERNZERHOF M. Rationale for mixing exact exchange with density functional approximations. Chemical Physics, 1996,105(22):9982-9985. |
| [20] | PENG L, WANG L, NIE Y, et al. Dual-ligand synergistic modulation: a satisfactory strategy for simultaneously improving the activity and stability of oxygen evolution electrocatalysts. ACS Catalysis, 2017,7:8184-8191. |
| [21] | XUE Z, ZHANG X Y, QIN J Q, et al. Revealing Ni-based layered double hydroxides as high-efficiency electrocatalysts for the oxygen evolution reaction: a DFT study. Journal of Materials Chemistry A, 2019,7(40):23091-23097. |
| [22] | MURDACHAEW G, LAASONEN K. Oxygen evolution reaction on nitrogen-doped defective carbon nanotubes and graphene. Journal of Physical Chemistry C, 2018,122(45):25882-25892. |
| [23] | WANG L X, WU M H, LANG X Y,et al. High-performance nitrogen fixation over Mo atom modified defective α-MnO2(001). ChemCatChem, 2020,12:3937-3945. |
| [24] | GAO X P, ZHOU Y N, TAN Y J, et al. Graphyne doped with transition-metal single atoms as effective bifunctional electrocatalysts for water splitting. Applied Surface Science, 2019,492:8-15. |
| [25] | LI A, FENG J R, ZHANG Y, et al. Controllable tuning of Fe-N nanosheets by Co substitution for enhanced oxygen evolution reaction. Nano Energy, 2019,57:644-652. |
| [26] | HEESE-GARTLEIN J, RABE A, BEHRENS M. Challenges in the application of manganese oxide powders as OER electrocatalysts: synthesis, characterization, activity and stability of nine different Mn xOy compounds. Zeitschrift Für Anorganische und Allgemeine Chemie, 2021,647:1-11. |
| [27] | ZHOU Y N, GAO G P, Li Y, et al. Transition-metal single atoms in nitrogen-doped graphenes as efficient active centers for water splitting: a theoretical study. Physical Chemistry Chemical Physics, 2019,21(6):7959-7966. |
| [28] | LI R C, HU B H, YU T W, et al. Insights into correlation among surface-structure-activity of cobalt-derived pre-catalyst for oxygen evolution reaction. Advanced Science, 2020,7(5):1902830. |
| [29] | LEE T H, LEE S A, PARK H,et al. Understanding the enhancement of the catalytic properties of goethite by transition metal doping: critical role of O* formation energy relative to OH* and OOH*. ACS Applied Energy Materials, 2020,3(2):1634-1643. |
| [1] | LIU Lei, GUO Ruihua, WANG Li, WANG Yan, ZHANG Guofang, GUAN Lili. Oxygen Reduction Reaction on Pt3Co High-index Facets by Density Functional Theory [J]. Journal of Inorganic Materials, 2025, 40(1): 39-46. |
| [2] | LI Jiaqi, LI Xiaosong, LI Xuanhe, ZHU Xiaobing, ZHU Aimin. Transition Metal-doped Manganese Oxide: Synthesis by Warm Plasma and Electrocatalytic Performance for Oxygen Evolution Reaction [J]. Journal of Inorganic Materials, 2024, 39(7): 835-844. |
| [3] | LI Honglan, ZHANG Junmiao, SONG Erhong, YANG Xinglin. Mo/S Co-doped Graphene for Ammonia Synthesis: a Density Functional Theory Study [J]. Journal of Inorganic Materials, 2024, 39(5): 561-568. |
| [4] | WU Guangyu, SHU Song, ZHANG Hongwei, LI Jianjun. Enhanced Styrene Adsorption by Grafted Lactone-based Activated Carbon [J]. Journal of Inorganic Materials, 2024, 39(4): 390-398. |
| [5] | YUE Quanxin, GUO Ruihua, WANG Ruifen, AN Shengli, ZHANG Guofang, GUAN Lili. 3D Core-shell Structured NiMoO4@CoFe-LDH Nanorods: Performance of Efficient Oxygen Evolution Reaction and Overall Water Splitting [J]. Journal of Inorganic Materials, 2024, 39(11): 1254-1264. |
| [6] | XIE Tian, SONG Erhong. Effect of Elastic Strains on Adsorption Energies of C, H and O on Transition Metal Oxides [J]. Journal of Inorganic Materials, 2024, 39(11): 1292-1300. |
| [7] | FU Yongsheng, BI Min, LI Chun, SUN Jingwen, WANG Xin, ZHU Junwu. Research Progress on Non-noble Metal/Nitrogen-doped Carbon Composite Materials in Electrocatalytic Oxygen Evolution Reaction [J]. Journal of Inorganic Materials, 2022, 37(2): 163-172. |
| [8] | WU Jing, YU Libing, LIU Shuaishuai, HUANG Qiuyan, JIANG Shanshan, ANTON Matveev, WANG Lianli, SONG Erhong, XIAO Beibei. NiN4/Cr Embedded Graphene for Electrochemical Nitrogen Fixation [J]. Journal of Inorganic Materials, 2022, 37(10): 1141-1148. |
| [9] | CAI Jianfeng, WANG Hongxiang, LIU Guoqiang, JIANG Jun. Designing High Entropy Structure in Thermoelectrics [J]. Journal of Inorganic Materials, 2021, 36(4): 399-404. |
| [10] | ZHANG Ruihong, WEI Xin, LU Zhanhui, AI Yuejie. Training Model for Predicting Adsorption Energy of Metal Ions Based on Machine Learning [J]. Journal of Inorganic Materials, 2021, 36(11): 1178-1184. |
| [11] | HE Junlong, SONG Erhong, WANG Lianjun, JIANG Wan. DFT Calculation of NO Adsorption on Cr Doped Graphene [J]. Journal of Inorganic Materials, 2021, 36(10): 1047-1052. |
| [12] | ZHOU Zihang, WANG Qun, GE Xiang, LI Zhaoyang. Strontium Doped Hydroxyapatite Nanoparticles: Synthesis, Characterization and Simulation [J]. Journal of Inorganic Materials, 2020, 35(11): 1283-1289. |
| [13] | QI Xin-Xin, SONG Guang-Ping, YIN Wei-Long, WANG Ming-Fu, HE Xiao-Dong, ZHENG Yong-Ting, WANG Rong-Guo, BAI Yue-Lei. Analysis on Phase Stability and Mechanical Property of Newly-discovered Ternary Layered Boride Cr4AlB4 [J]. Journal of Inorganic Materials, 2020, 35(1): 53-60. |
| [14] | LI Yuan-Yang, JIANG Bo. λ/4-λ/2 Double-layer Broadband Antireflective Coatings with Superhydrophilicity and Photocatalysis [J]. Journal of Inorganic Materials, 2019, 34(2): 159-163. |
| [15] | WANG Jun-Kai, ZHANG Yuan-Zhuo, LI Jun-Yi, ZHANG Hai-Jun, LI Fa-Liang, HAN Lei, SONG Shu-Peng. Low Temperature Catalytic Synthesis of β-SiC Powders via Microwave Heating [J]. Journal of Inorganic Materials, 2017, 32(7): 725-730. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||